

Long-read whole-genome analysis of human single cells
- PMID: 37620373
- PMCID: PMC10449900
- DOI: 10.1038/s41467-023-40898-3
Long-read whole-genome analysis of human single cells
Abstract
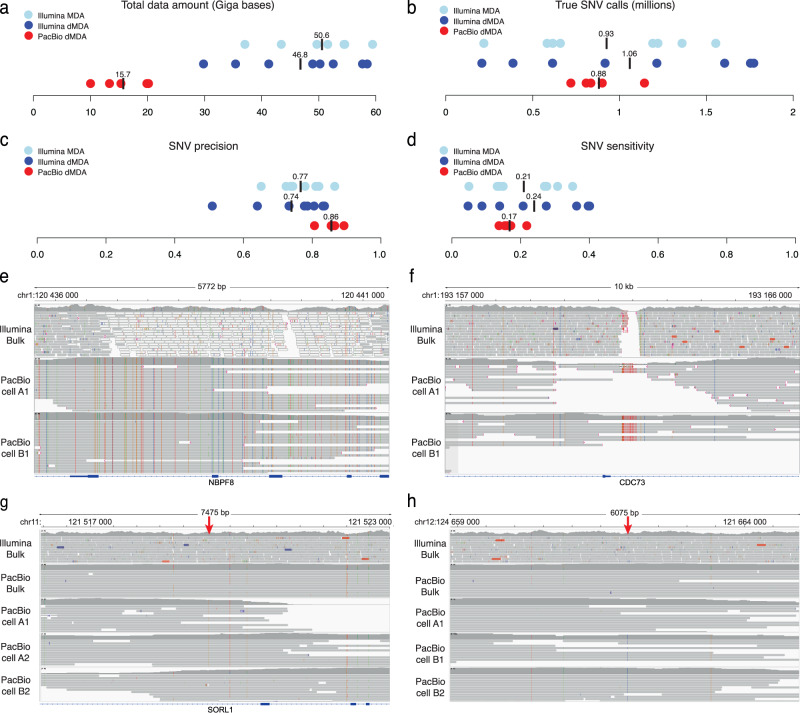
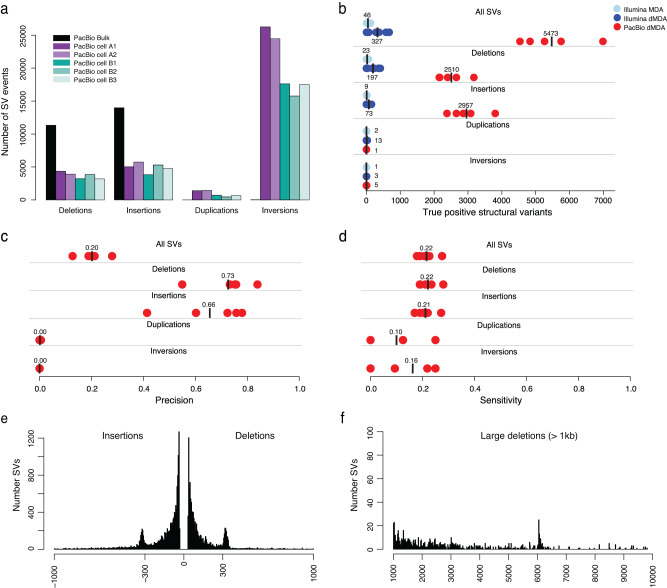
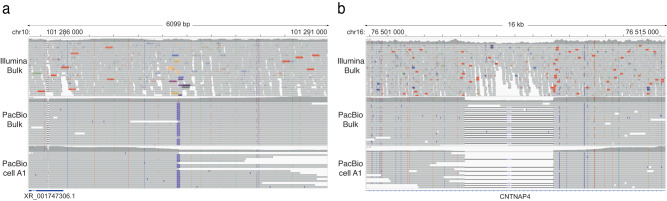
Long-read sequencing has dramatically increased our understanding of human genome variation. Here, we demonstrate that long-read technology can give new insights into the genomic architecture of individual cells. Clonally expanded CD8+ T-cells from a human donor were subjected to droplet-based multiple displacement amplification (dMDA) to generate long molecules with reduced bias. PacBio sequencing generated up to 40% genome coverage per single-cell, enabling detection of single nucleotide variants (SNVs), structural variants (SVs), and tandem repeats, also in regions inaccessible by short reads. 28 somatic SNVs were detected, including one case of mitochondrial heteroplasmy. 5473 high-confidence SVs/cell were discovered, a sixteen-fold increase compared to Illumina-based results from clonally related cells. Single-cell de novo assembly generated a genome size of up to 598 Mb and 1762 (12.8%) complete gene models. In summary, our work shows the promise of long-read sequencing toward characterization of the full spectrum of genetic variation in single cells.
© 2023. Springer Nature Limited.
Conflict of interest statement
C.-S.C. is an employee and shareholder of GeneDX, LLC. The other authors declare no competing interests. Parts of the sequencing costs were funded by Samplix.
Figures



References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
