

Multiparameter prediction of myeloid neoplasia risk
- PMID: 37620601
- PMCID: PMC10484784
- DOI: 10.1038/s41588-023-01472-1
Multiparameter prediction of myeloid neoplasia risk
Erratum in
-
Author Correction: Multiparameter prediction of myeloid neoplasia risk.Nat Genet. 2023 Oct;55(10):1777. doi: 10.1038/s41588-023-01532-6. Nat Genet. 2023. PMID: 37726541 Free PMC article. No abstract available.
Abstract
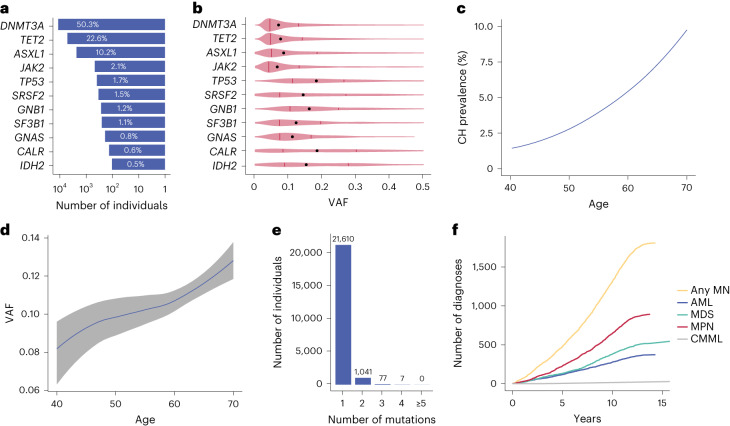
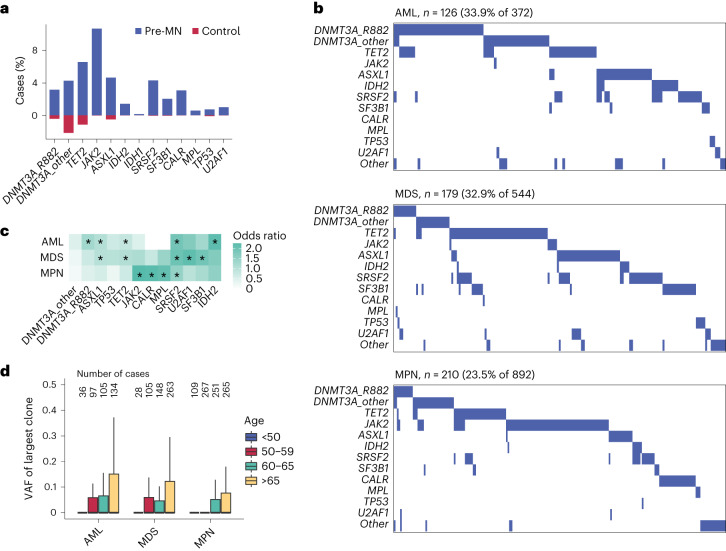
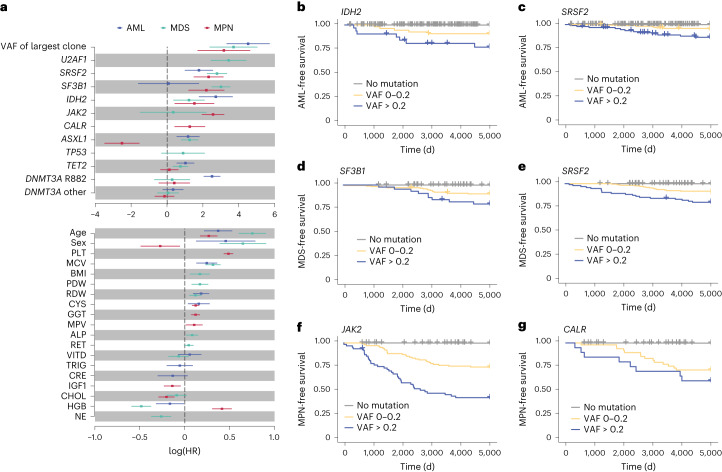
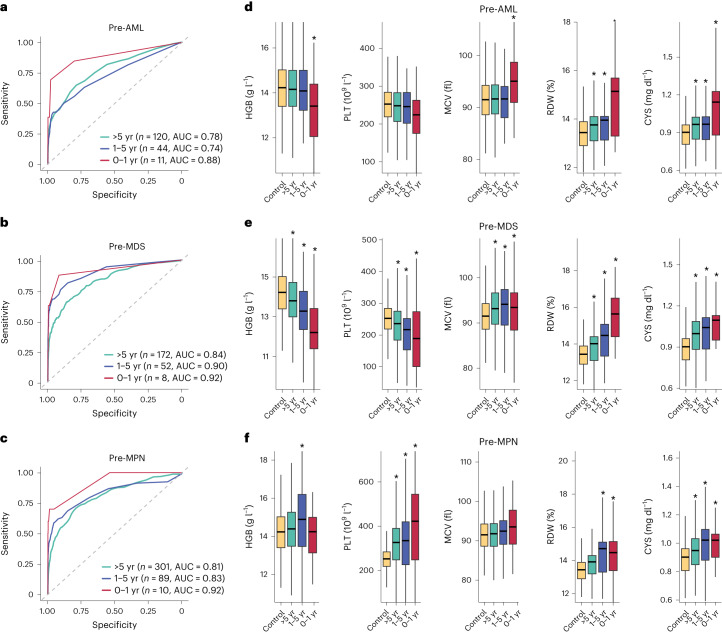
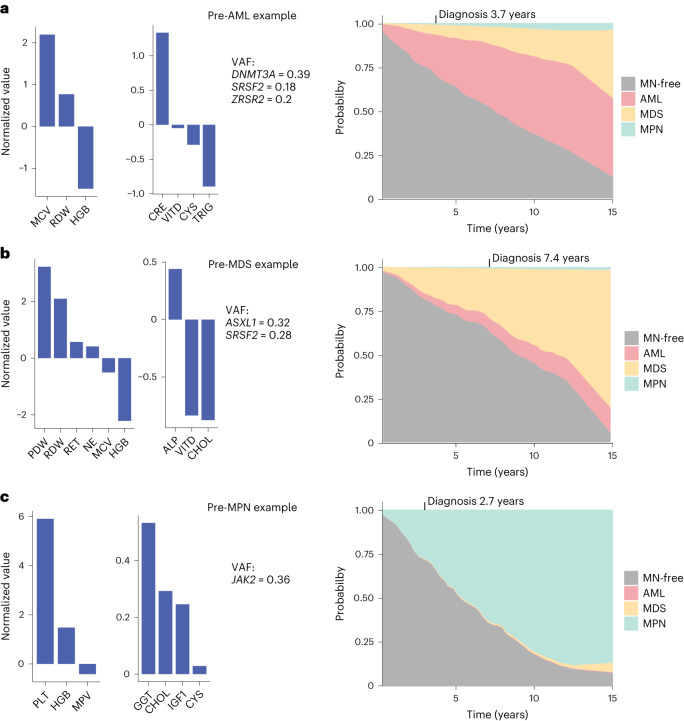
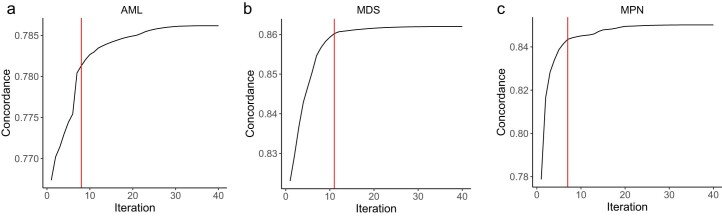
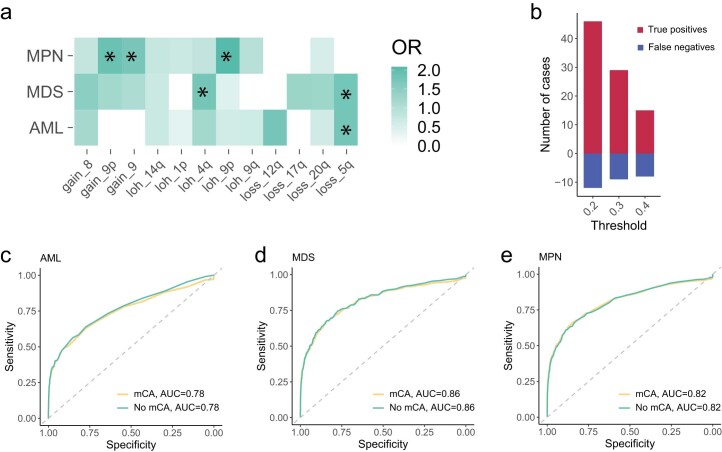
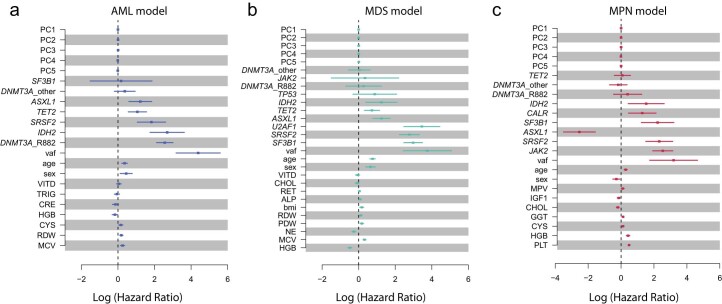
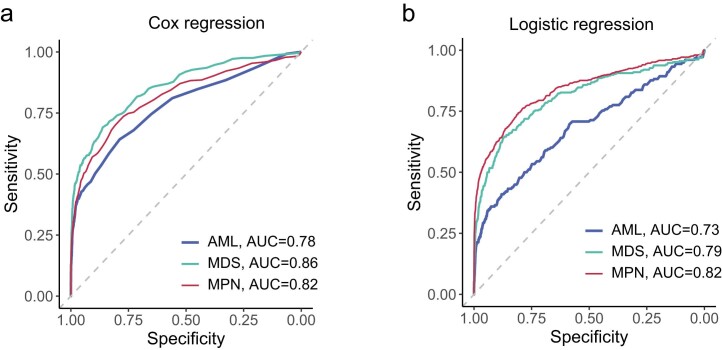
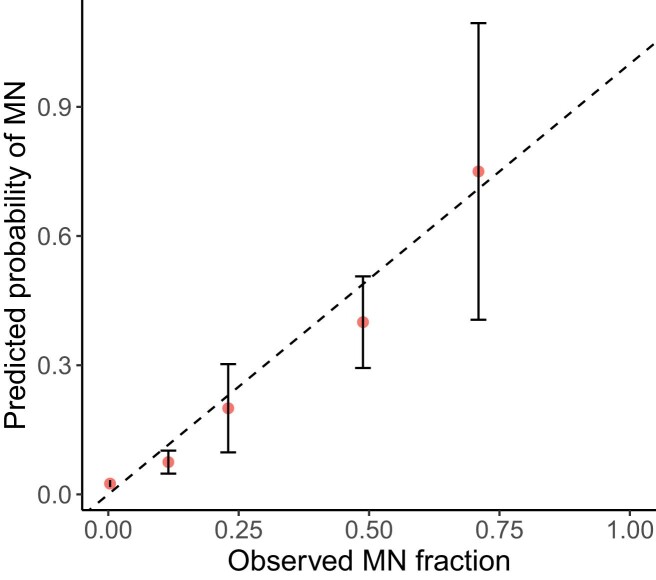
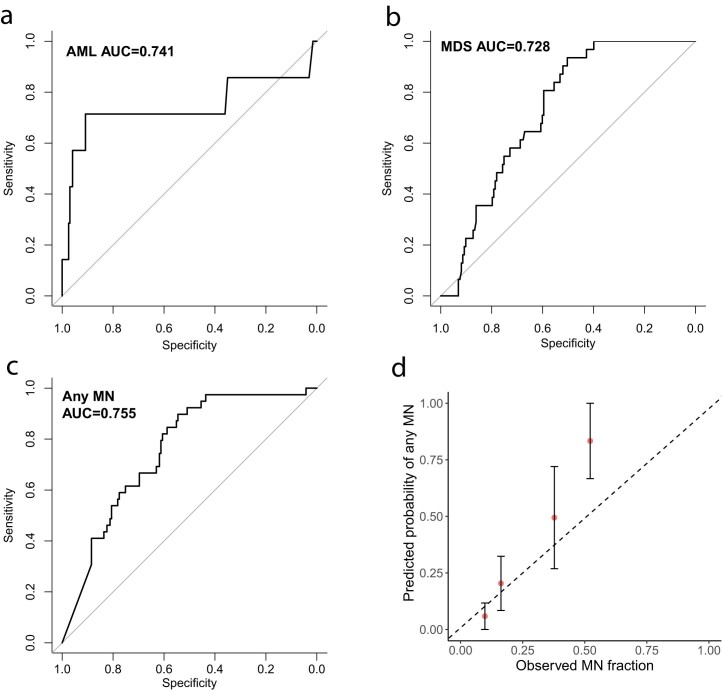
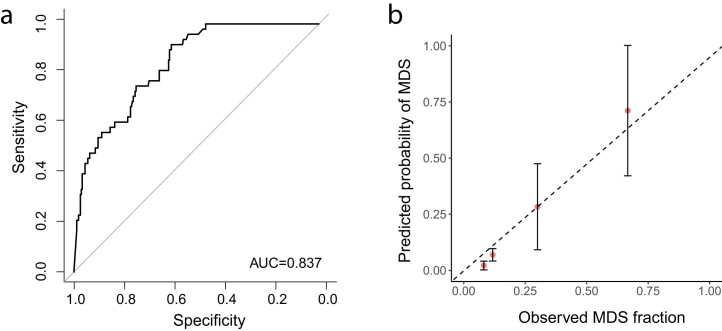
The myeloid neoplasms encompass acute myeloid leukemia, myelodysplastic syndromes and myeloproliferative neoplasms. Most cases arise from the shared ancestor of clonal hematopoiesis (CH). Here we analyze data from 454,340 UK Biobank participants, of whom 1,808 developed a myeloid neoplasm 0-15 years after recruitment. We describe the differences in CH mutational landscapes and hematology/biochemistry test parameters among individuals that later develop myeloid neoplasms (pre-MN) versus controls, finding that disease-specific changes are detectable years before diagnosis. By analyzing differences between 'pre-MN' and controls, we develop and validate Cox regression models quantifying the risk of progression to each myeloid neoplasm subtype. We construct 'MN-predict', a web application that generates time-dependent predictions with the input of basic blood tests and genetic data. Our study demonstrates that many individuals that develop myeloid neoplasms can be identified years in advance and provides a framework for disease-specific prognostication that will be of substantial use to researchers and physicians.
© 2023. The Author(s).
Conflict of interest statement
G.S.V. is a consultant to STRM.BIO and holds a research grant from AstraZeneca for research unrelated to that presented here. M.A.F. is an employee and stockholder of AstraZeneca. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
