

The Urine Metabolome of R6/2 and zQ175DN Huntington's Disease Mouse Models
- PMID: 37623904
- PMCID: PMC10456449
- DOI: 10.3390/metabo13080961
The Urine Metabolome of R6/2 and zQ175DN Huntington's Disease Mouse Models
Abstract
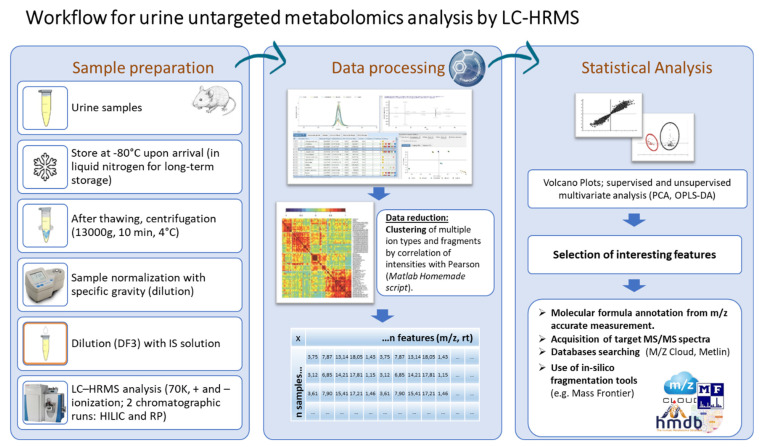
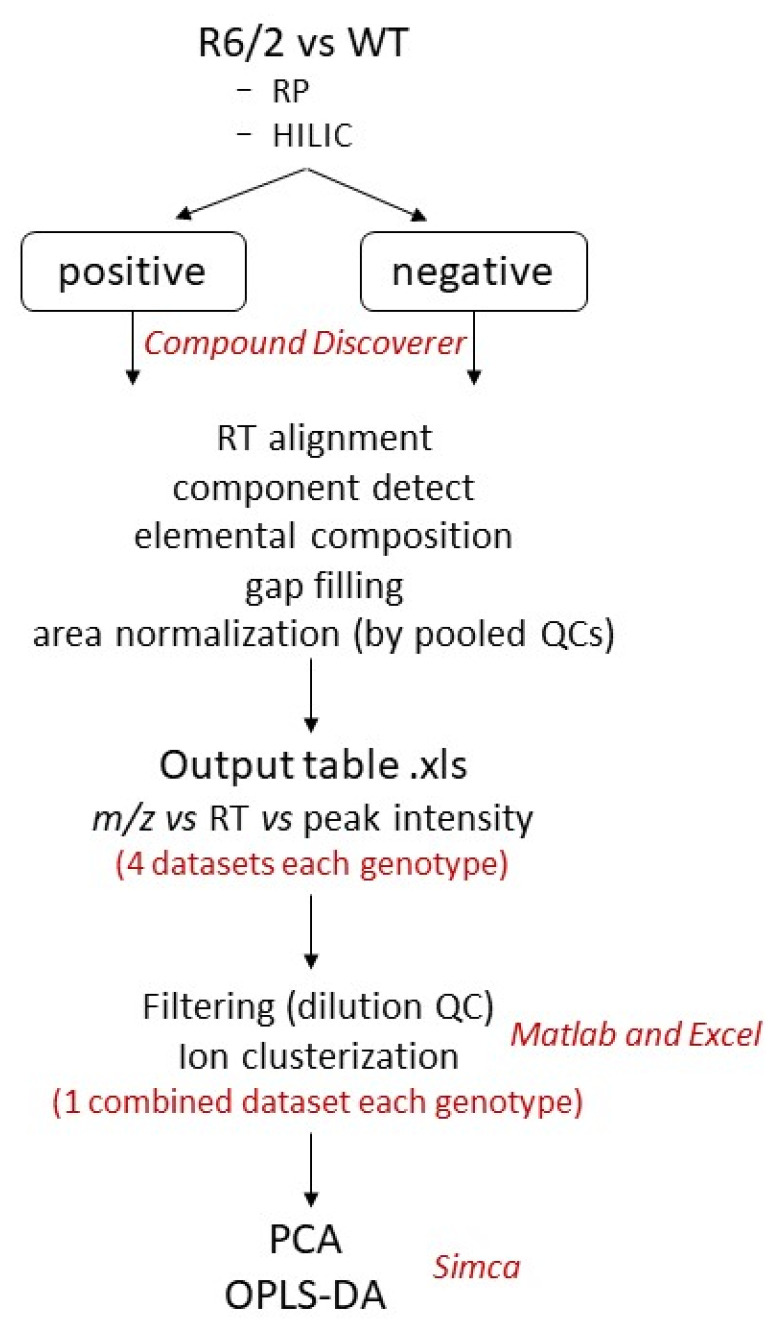
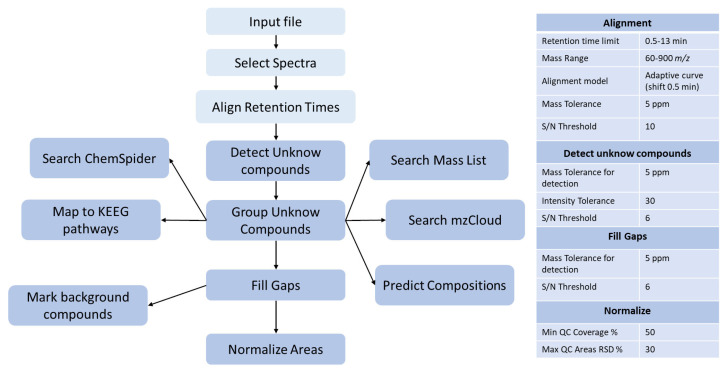
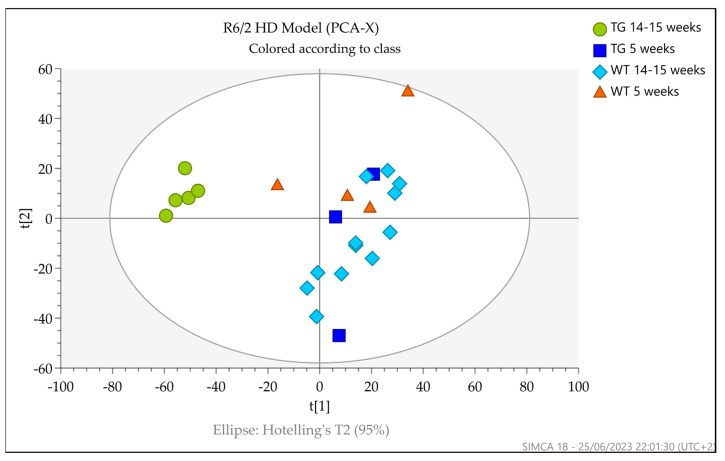
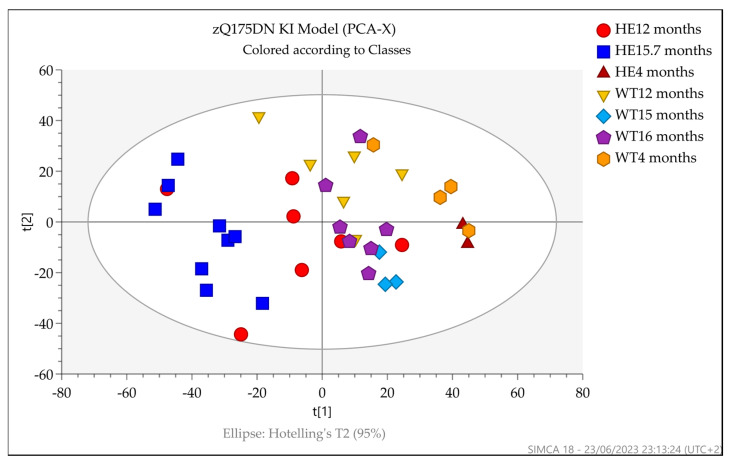
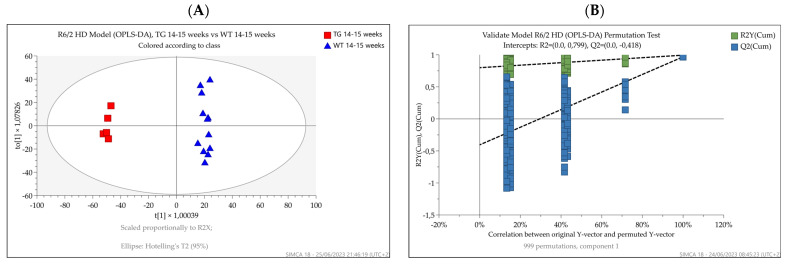
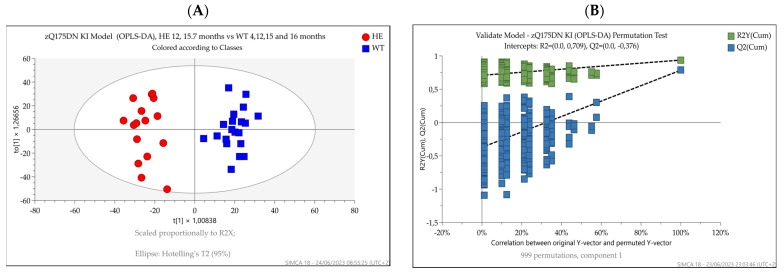
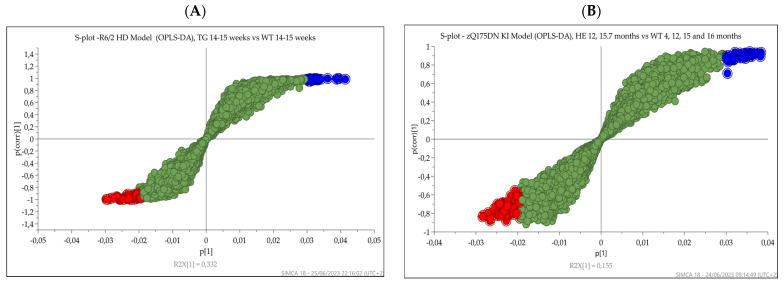
Huntington's disease (HD) is caused by the expansion of a polyglutamine (polyQ)-encoding tract in exon 1 of the huntingtin gene to greater than 35 CAG repeats. It typically has a disease course lasting 15-20 years, and there are currently no disease-modifying therapies available. Thus, there is a need for faithful mouse models of HD to use in preclinical studies of disease mechanisms, target validation, and therapeutic compound testing. A large variety of mouse models of HD were generated, none of which fully recapitulate human disease, complicating the selection of appropriate models for preclinical studies. Here, we present the urinary liquid chromatography-high-resolution mass spectrometry analysis employed to identify metabolic alterations in transgenic R6/2 and zQ175DN knock-in mice. In R6/2 mice, the perturbation of the corticosterone metabolism and the accumulation of pyrraline, indicative of the development of insulin resistance and the impairment of pheromone excretion, were observed. Differently from R6/2, zQ175DN mice showed the accumulation of oxidative stress metabolites. Both genotypes showed alterations in the tryptophan metabolism. This approach aims to improve our understanding of the molecular mechanisms involved in HD neuropathology, facilitating the selection of appropriate mouse models for preclinical studies. It also aims to identify potential biomarkers specific to HD.
Keywords: Huntington’s disease; R6/2 mice; mass spectrometry; urine metabolome; zQ175DN mice.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Formation of polyglutamine inclusions in a wide range of non-CNS tissues in the HdhQ150 knock-in mouse model of Huntington's disease.PLoS One. 2009 Nov 30;4(11):e8025. doi: 10.1371/journal.pone.0008025. PLoS One. 2009. PMID: 19956633 Free PMC article.
-
Normalizing glucocorticoid levels attenuates metabolic and neuropathological symptoms in the R6/2 mouse model of huntington's disease.Neurobiol Dis. 2019 Jan;121:214-229. doi: 10.1016/j.nbd.2018.09.025. Epub 2018 Oct 5. Neurobiol Dis. 2019. PMID: 30292559 Free PMC article.
-
Distinct histological alterations of cortical interneuron types in mouse models of Huntington's disease.Front Neurosci. 2022 Sep 26;16:1022251. doi: 10.3389/fnins.2022.1022251. eCollection 2022. Front Neurosci. 2022. PMID: 36225731 Free PMC article.
-
Huntington's disease mouse models: unraveling the pathology caused by CAG repeat expansion.Fac Rev. 2021 Oct 21;10:77. doi: 10.12703/r/10-77. eCollection 2021. Fac Rev. 2021. PMID: 34746930 Free PMC article. Review.
-
The contribution of preclinical magnetic resonance imaging and spectroscopy to Huntington's disease.Front Aging Neurosci. 2024 Feb 13;16:1306312. doi: 10.3389/fnagi.2024.1306312. eCollection 2024. Front Aging Neurosci. 2024. PMID: 38414634 Free PMC article. Review.
Cited by
-
Potential molecular mechanism of exercise reversing insulin resistance and improving neurodegenerative diseases.Front Physiol. 2024 May 16;15:1337442. doi: 10.3389/fphys.2024.1337442. eCollection 2024. Front Physiol. 2024. PMID: 38818523 Free PMC article. Review.
-
Tackling new psychoactive substances through metabolomics: UHPLC-HRMS study on natural and synthetic opioids in male and female murine models.Sci Rep. 2024 Apr 24;14(1):9432. doi: 10.1038/s41598-024-60045-2. Sci Rep. 2024. PMID: 38658766 Free PMC article.
References
-
- Bates G.P., Jones L. Huntington’s Disease. Oxford University Press; Oxford, UK: 2002.
-
- Rubinsztein D.C., Leggo J., Coles R., Almqvist E., Biancalana V., Cassiman J.J., Chorai K., Connarty M., Crauford D., Curtis A., et al. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am. J. Hum. Genet. 1996;59:16–22. - PMC - PubMed
LinkOut - more resources
Full Text Sources
