

Novel α-1,3-Glucosyltransferase Variants and Their Broad Clinical Polycystic Liver Disease Spectrum
- PMID: 37628703
- PMCID: PMC10454741
- DOI: 10.3390/genes14081652
Novel α-1,3-Glucosyltransferase Variants and Their Broad Clinical Polycystic Liver Disease Spectrum
Abstract
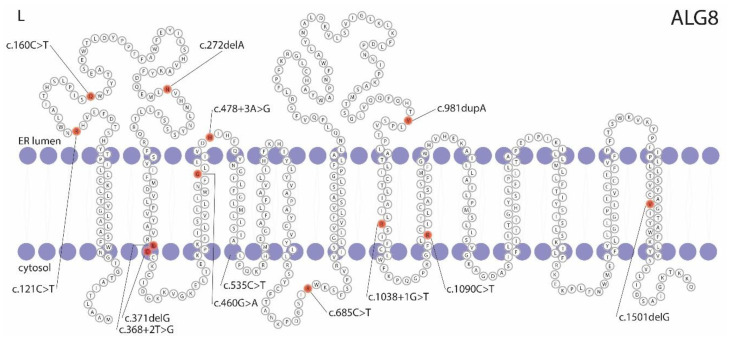
Protein-truncating variants in α-1,3-glucosyltransferase (ALG8) are a risk factor for a mild cystic kidney disease phenotype. The association between these variants and liver cysts is limited. We aim to identify pathogenic ALG8 variants in our cohort of autosomal dominant polycystic liver disease (ADPLD) individuals. In order to fine-map the phenotypical spectrum of pathogenic ALG8 variant carriers, we performed targeted ALG8 screening in 478 ADPLD singletons, and exome sequencing in 48 singletons and 4 patients from two large ADPLD families. Eight novel and one previously reported pathogenic variant in ALG8 were discovered in sixteen patients. The ALG8 clinical phenotype ranges from mild to severe polycystic liver disease, and from innumerable small to multiple large hepatic cysts. The presence of <5 renal cysts that do not affect renal function is common in this population. Three-dimensional homology modeling demonstrated that six variants cause a truncated ALG8 protein with abnormal functioning, and one variant is predicted to destabilize ALG8. For the seventh variant, immunostaining of the liver tissue showed a complete loss of ALG8 in the cystic cells. ALG8-associated ADPLD has a broad clinical spectrum, including the possibility of developing a small number of renal cysts. This broadens the ADPLD genotype-phenotype spectrum and narrows the gap between liver-specific ADPLD and kidney-specific ADPKD.
Keywords: ADPLD; ALG8; clinical spectrum; next-generation sequencing; polycystic liver disease.
Conflict of interest statement
J.P.H.D. declares that the Radboudumc, on behalf of J.P.H.D., received honoraria or research grants from Gilead, and J.P.H.D. is the PI of the POSITANO study (Camurus). The other authors declare no conflicts of interest. The funder, RIMLS/RIHI, had no role in the design of the study and the collection, analysis, and interpretation of the data.
Figures




Similar articles
-
Heterozygosity of ALG9 in Association with Autosomal Dominant Polycystic Liver Disease.Genes (Basel). 2023 Sep 2;14(9):1755. doi: 10.3390/genes14091755. Genes (Basel). 2023. PMID: 37761895 Free PMC article.
-
Prevalence Estimates of Polycystic Kidney and Liver Disease by Population Sequencing.J Am Soc Nephrol. 2018 Oct;29(10):2593-2600. doi: 10.1681/ASN.2018050493. Epub 2018 Aug 22. J Am Soc Nephrol. 2018. PMID: 30135240 Free PMC article.
-
Individuals heterozygous for ALG8 protein-truncating variants are at increased risk of a mild cystic kidney disease.Kidney Int. 2023 Mar;103(3):607-615. doi: 10.1016/j.kint.2022.11.025. Epub 2022 Dec 24. Kidney Int. 2023. PMID: 36574950 Free PMC article.
-
Polycystic liver disease genes: Practical considerations for genetic testing.Eur J Med Genet. 2021 Mar;64(3):104160. doi: 10.1016/j.ejmg.2021.104160. Epub 2021 Feb 6. Eur J Med Genet. 2021. PMID: 33556586 Review.
-
Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases.J Am Soc Nephrol. 2018 Jan;29(1):13-23. doi: 10.1681/ASN.2017050483. Epub 2017 Oct 16. J Am Soc Nephrol. 2018. PMID: 29038287 Free PMC article. Review.
Cited by
-
Heterozygosity of ALG9 in Association with Autosomal Dominant Polycystic Liver Disease.Genes (Basel). 2023 Sep 2;14(9):1755. doi: 10.3390/genes14091755. Genes (Basel). 2023. PMID: 37761895 Free PMC article.
-
Characterization of the Cystic Phenotype Associated with Monoallelic ALG8 and ALG9 Pathogenic Variants.J Am Soc Nephrol. 2025 Jun 1;36(6):1056-1071. doi: 10.1681/ASN.0000000613. Epub 2025 Feb 3. J Am Soc Nephrol. 2025. PMID: 39899384
References
-
- Olaizola P., Rodrigues P.M., Caballero-Camino F.J., Izquierdo-Sanchez L., Aspichueta P., Bujanda L., Larusso N.F., Drenth J.P.H., Perugorria M.J., Banales J.M. Genetics, pathobiology and therapeutic opportunities of polycystic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2022;19:585–604. doi: 10.1038/s41575-022-00617-7. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
