

Current insights in the molecular genetic pathogenesis of amyotrophic lateral sclerosis
- PMID: 37638324
- PMCID: PMC10448825
- DOI: 10.3389/fnins.2023.1189470
Current insights in the molecular genetic pathogenesis of amyotrophic lateral sclerosis
Abstract
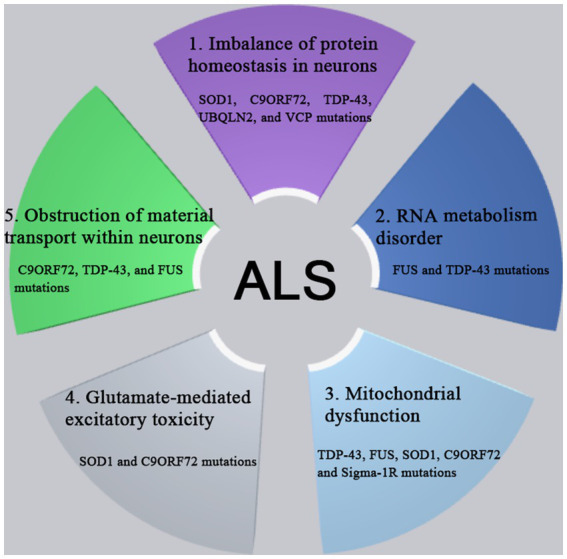
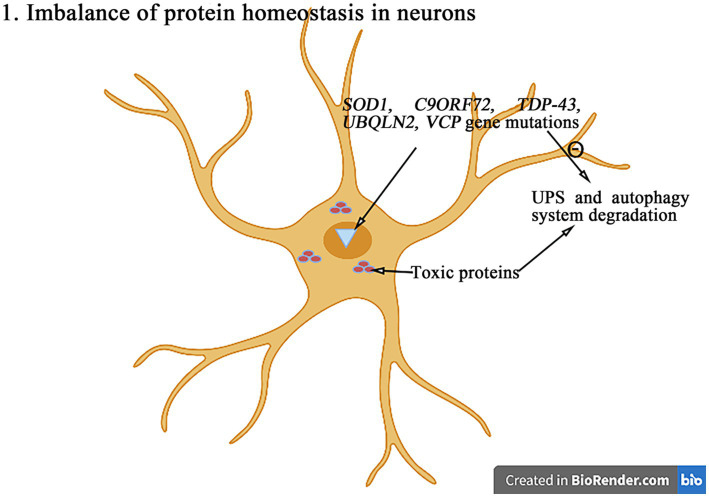
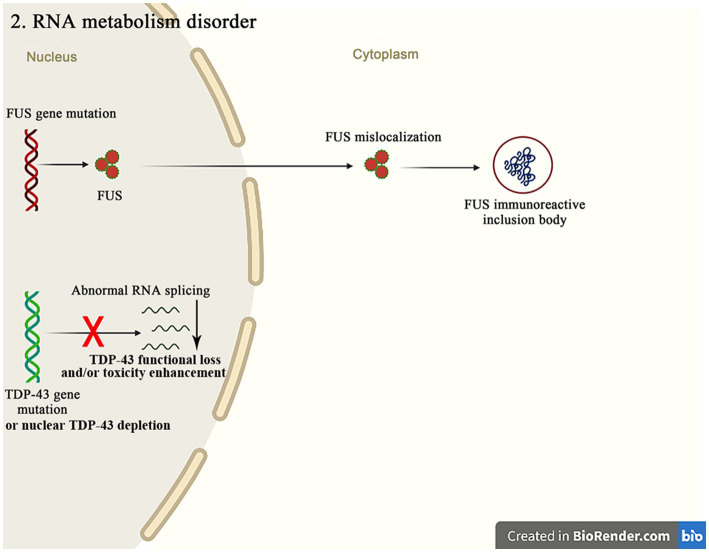
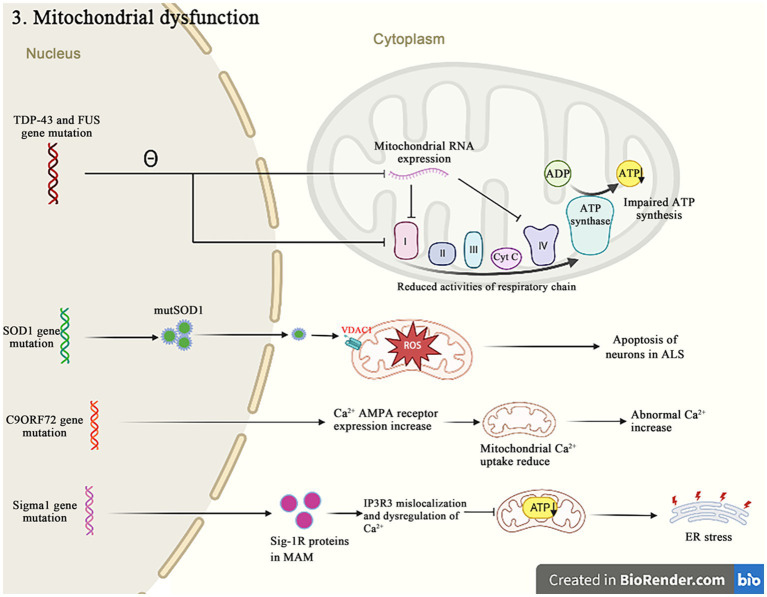
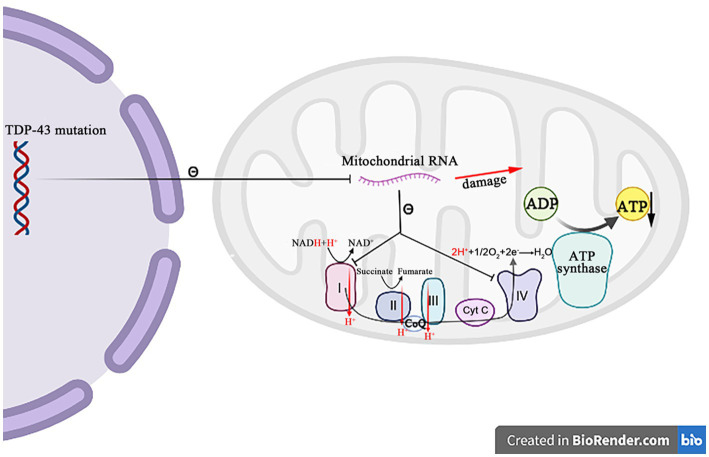
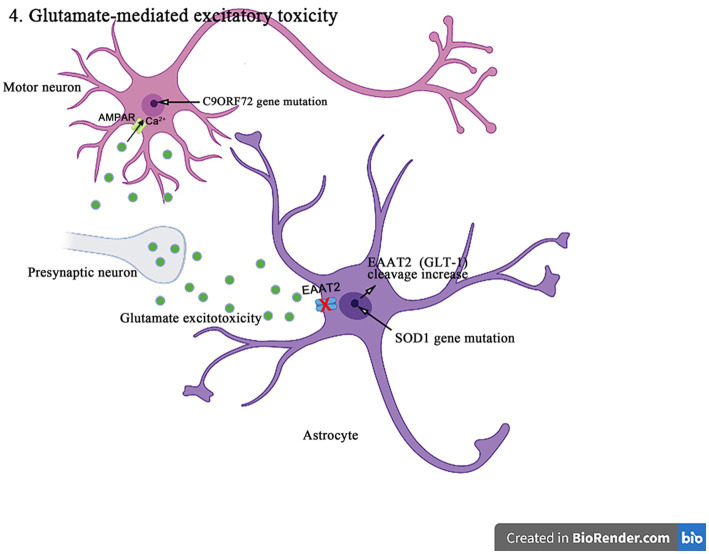
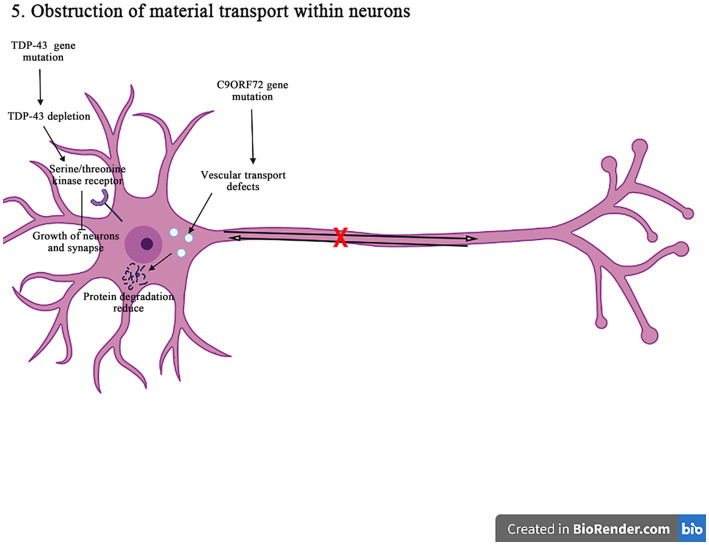
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease that leads to the massive loss of motor neurons in cerebrum, brain stem and spinal cord. It affects not only motor neurons but also other neurons and glial cells, resulting in the progressive muscle atrophy, the severe disability and the eventual death due to the respiratory failure. The pathogenesis of ALS is not fully understood. Currently, several factors are considered to be involved in the pathogenesis of ALS, such as genetic factors, imbalances in protein homeostasis, RNA metabolism disorders, mitochondrial dysfunctions, glutamate-mediated excitatory toxicities and intra-neuronal material transport disorders in neurons. The study of genetic mutations related to ALS pathogenesis will link the molecular and cellular mechanisms of the disease, thus enhancing the understanding of its occurrence and progression, thereby providing new insights for the pathogenesis of ALS. This review summarizes the current insights in the molecular genetic pathogenesis of ALS.
Keywords: amyotrophic lateral sclerosis; genetics; molecule; pathogenesis; proteomics.
Copyright © 2023 Zhou and Xu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Advances in genetics research in the pathogenesis of amyotrophic lateral sclerosis.Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2020 Dec 28;45(12):1483-1489. doi: 10.11817/j.issn.1672-7347.2020.190506. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2020. PMID: 33473007 Chinese, English.
-
Single-cell RNA-seq analysis of the brainstem of mutant SOD1 mice reveals perturbed cell types and pathways of amyotrophic lateral sclerosis.Neurobiol Dis. 2020 Jul;141:104877. doi: 10.1016/j.nbd.2020.104877. Epub 2020 Apr 30. Neurobiol Dis. 2020. PMID: 32360664 Free PMC article.
-
Macrophage-mediated inflammation and glial response in the skeletal muscle of a rat model of familial amyotrophic lateral sclerosis (ALS).Exp Neurol. 2016 Mar;277:275-282. doi: 10.1016/j.expneurol.2016.01.008. Epub 2016 Jan 13. Exp Neurol. 2016. PMID: 26775178 Free PMC article.
-
Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments.Front Neurosci. 2017 Jan 10;10:611. doi: 10.3389/fnins.2016.00611. eCollection 2016. Front Neurosci. 2017. PMID: 28119559 Free PMC article. Review.
-
Advances and challenges in understanding the multifaceted pathogenesis of amyotrophic lateral sclerosis.Swiss Med Wkly. 2015 Jan 30;145:w14054. doi: 10.4414/smw.2015.14054. eCollection 2015. Swiss Med Wkly. 2015. PMID: 25635517 Review.
Cited by
-
Astrocyte-Neuron Interactions Contributing to Amyotrophic Lateral Sclerosis Progression.Adv Neurobiol. 2024;39:285-318. doi: 10.1007/978-3-031-64839-7_12. Adv Neurobiol. 2024. PMID: 39190080 Review.
-
Genetic Modifiers of ALS: The Impact of Chromogranin B P413L in a Bulgarian ALS Cohort.Genes (Basel). 2024 Sep 12;15(9):1197. doi: 10.3390/genes15091197. Genes (Basel). 2024. PMID: 39336788 Free PMC article.
-
Muscle Involvement in Amyotrophic Lateral Sclerosis: Understanding the Pathogenesis and Advancing Therapeutics.Biomolecules. 2023 Oct 26;13(11):1582. doi: 10.3390/biom13111582. Biomolecules. 2023. PMID: 38002264 Free PMC article. Review.
-
Superoxide dismutase activity in tear fluid and blood of patients and mouse model of amyotrophic lateral sclerosis: a pilot study.PeerJ. 2025 Jul 1;13:e19623. doi: 10.7717/peerj.19623. eCollection 2025. PeerJ. 2025. PMID: 40620778 Free PMC article.
-
Modulatory Effects of Urtica dioica on Neurodegenerative Diseases: Unveiling the Latest Findings and Applications Related to Neuroinflammation, Oxidative Stress, and Cognitive Dysfunction.Antioxidants (Basel). 2025 Jul 12;14(7):854. doi: 10.3390/antiox14070854. Antioxidants (Basel). 2025. PMID: 40722958 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous