

Unified Mouse and Human Kidney Single-Cell Expression Atlas Reveal Commonalities and Differences in Disease States
- PMID: 37639336
- PMCID: PMC10631616
- DOI: 10.1681/ASN.0000000000000217
Unified Mouse and Human Kidney Single-Cell Expression Atlas Reveal Commonalities and Differences in Disease States
Abstract
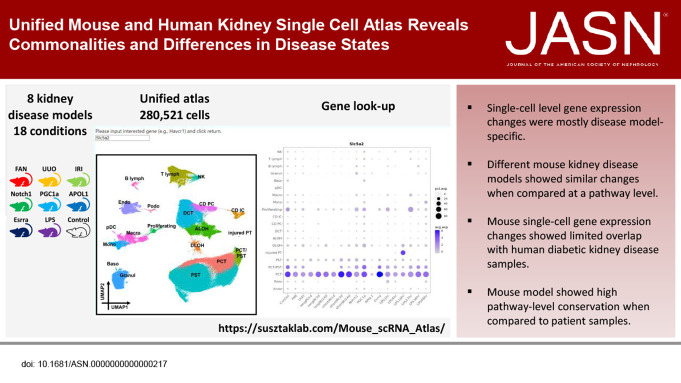
Significance statement: Mouse models have been widely used to understand kidney disease pathomechanisms and play an important role in drug discovery. However, these models have not been systematically analyzed and compared. The authors characterized 18 different mouse kidney disease models at both bulk and single-cell gene expression levels and compared single-cell gene expression data from diabetic kidney disease (DKD) mice and from patients with DKD. Although single cell-level gene expression changes were mostly model-specific, different disease models showed similar changes when compared at a pathway level. The authors also found that changes in fractions of cell types are major drivers of bulk gene expression differences. Although the authors found only a small overlap of single cell-level gene expression changes between the mouse DKD model and patients, they observed consistent pathway-level changes.
Background: Mouse models have been widely used to understand kidney disease pathomechanisms and play an important role in drug discovery. However, these models have not been systematically analyzed and compared.
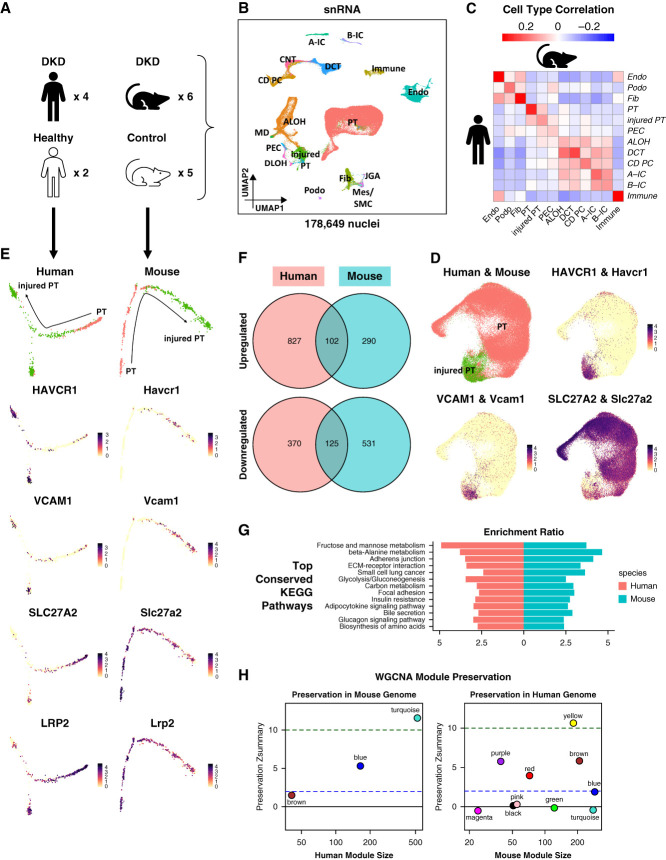
Methods: We analyzed single-cell RNA sequencing data (36 samples) and bulk gene expression data (42 samples) from 18 commonly used mouse kidney disease models. We compared single-nucleus RNA sequencing data from a mouse diabetic kidney disease model with data from patients with diabetic kidney disease and healthy controls.
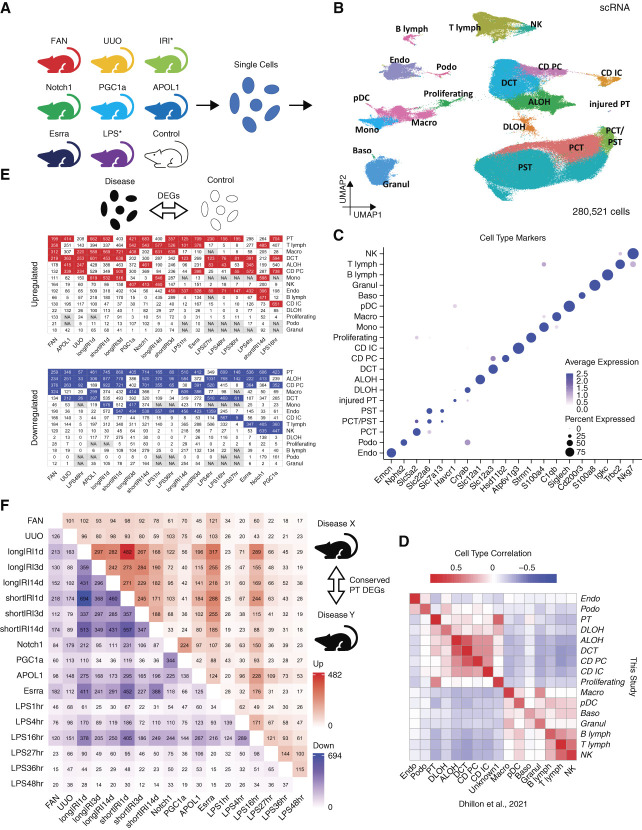
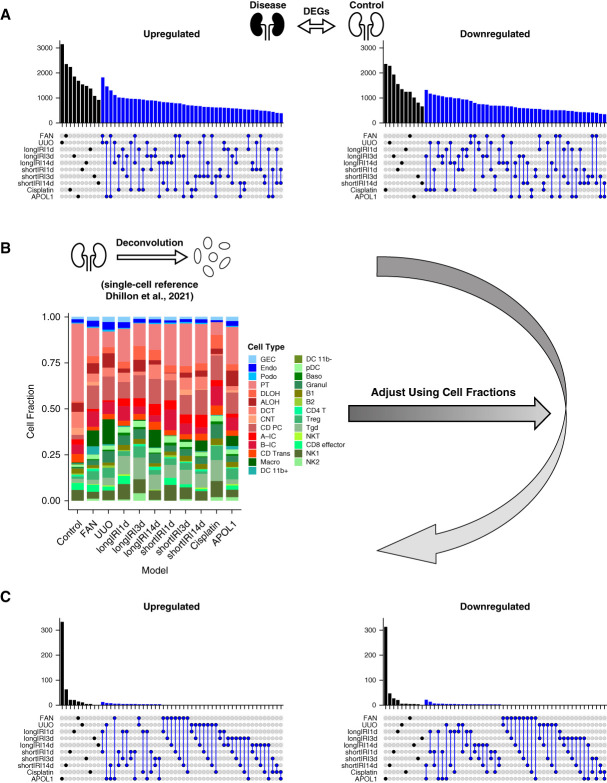
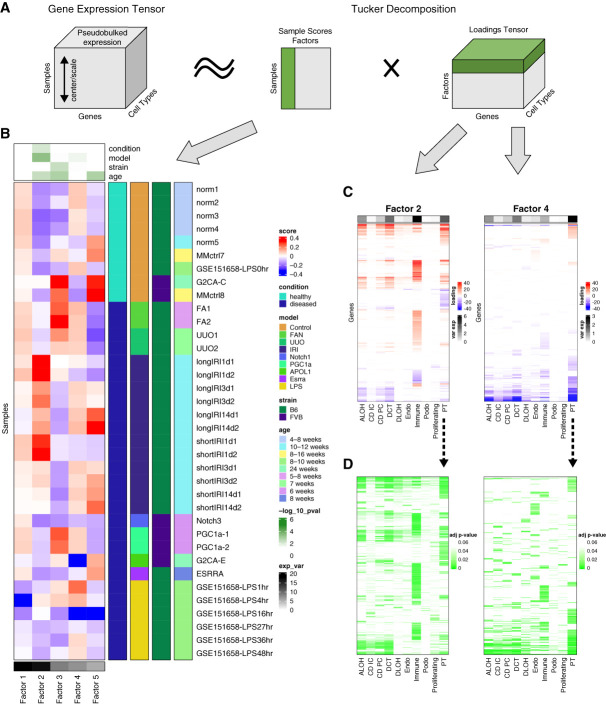
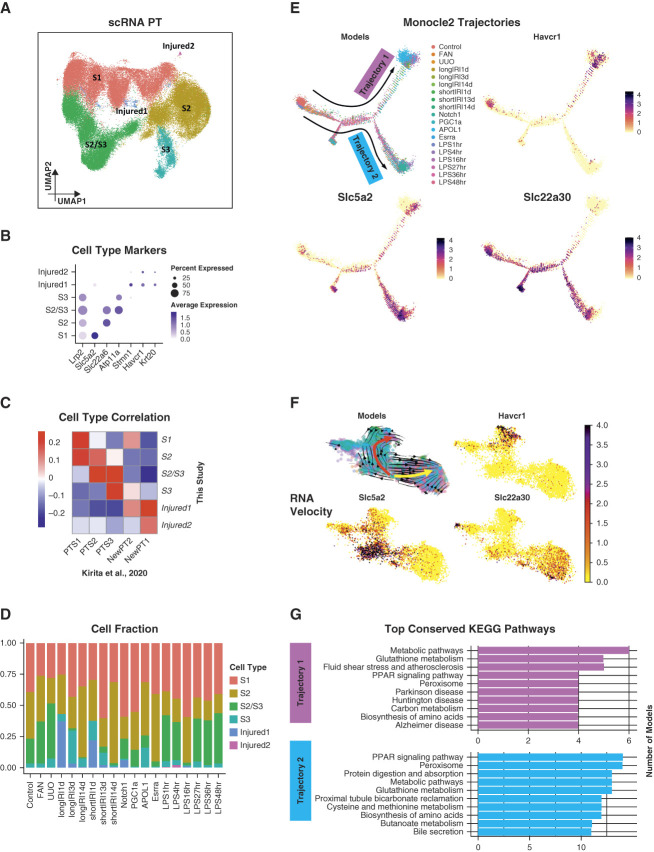
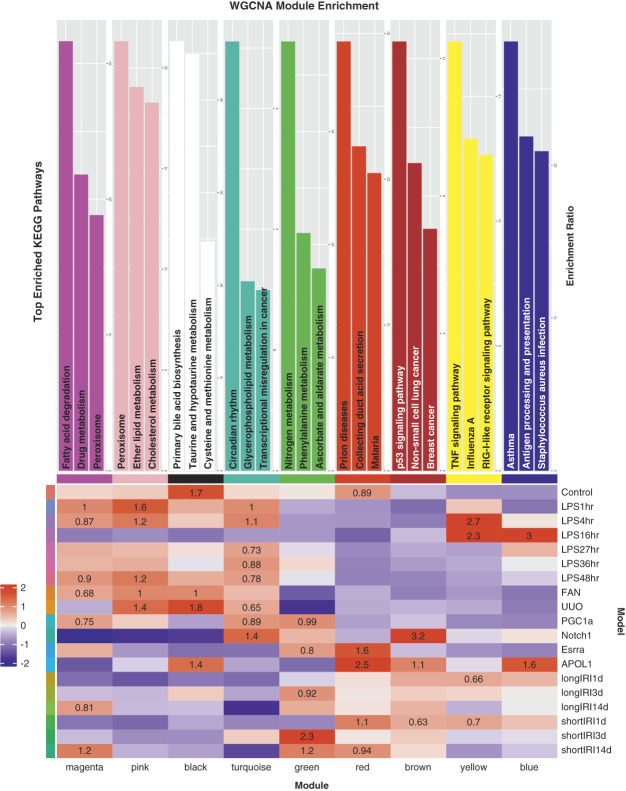
Results: We generated a uniformly processed mouse single-cell atlas containing information for nearly 300,000 cells, identifying all major kidney cell types and states. Our analysis revealed that changes in fractions of cell types are major drivers of differences in bulk gene expression. Although gene expression changes at the single-cell level were mostly model-specific, different disease models showed similar changes when compared at a pathway level. Tensor decomposition analysis highlighted the important changes in proximal tubule cells in disease states. Specifically, we identified important alterations in expression of metabolic and inflammation-associated pathways. The mouse diabetic kidney disease model and patients with diabetic kidney disease shared only a small number of conserved cell type-specific differentially expressed genes, but we observed pathway-level activation patterns conserved between mouse and human diabetic kidney disease samples.
Conclusions: This study provides a comprehensive mouse kidney single-cell atlas and defines gene expression commonalities and differences in disease states in mice. The results highlight the key role of cell heterogeneity in driving changes in bulk gene expression and the limited overlap of single-cell gene expression changes between animal models and patients, but they also reveal consistent pathway-level changes.
Copyright © 2023 by the American Society of Nephrology.
Conflict of interest statement
M.S. Balzer reports consultancy: Boehringer-Ingelheim; ownership interest: Arcturus Therapeutics, AstraZeneca, Bayer, BioNTech, CureVac, Linde, Moderna, Pfizer; honoraria: Boehringer-Ingelheim; and advisory or leadership role: Boehringer-Ingelheim,
Because Katalin Susztak is an editor of the
Figures
References
-
- National Kidney Foundation Research Roundtable Work Group on behalf of the National Kidney Foundation. Research priorities for kidney-related research-an agenda to advance kidney care: a position statement from the National kidney foundation. Am J Kidney Dis. 2022;79(2):141–152. doi:10.1053/j.ajkd.2021.08.018 - DOI - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
