

New mutation in the β1 propeller domain of LRP4 responsible for congenital myasthenic syndrome associated with Cenani-Lenz syndrome
- PMID: 37640745
- PMCID: PMC10462681
- DOI: 10.1038/s41598-023-41008-5
New mutation in the β1 propeller domain of LRP4 responsible for congenital myasthenic syndrome associated with Cenani-Lenz syndrome
Abstract
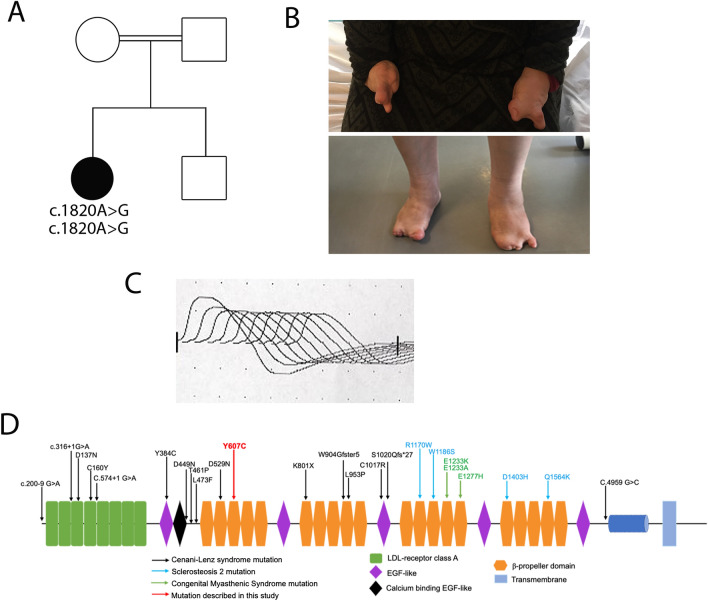
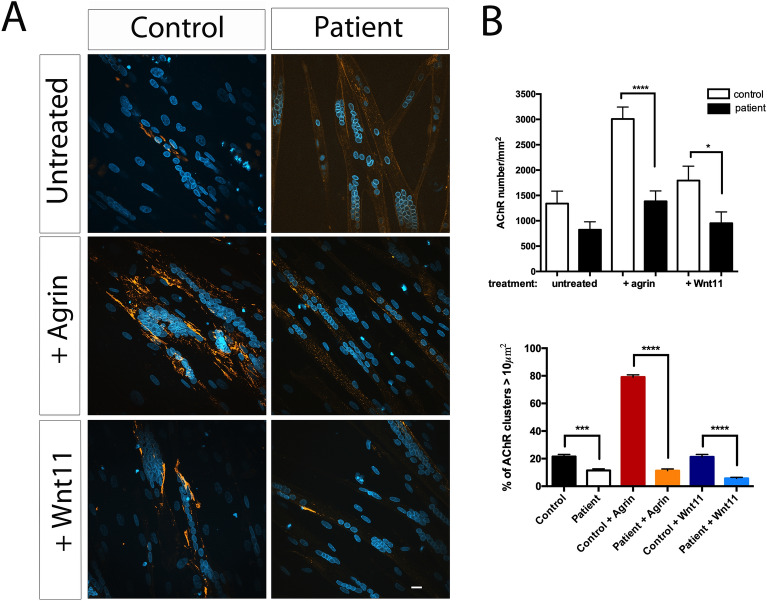
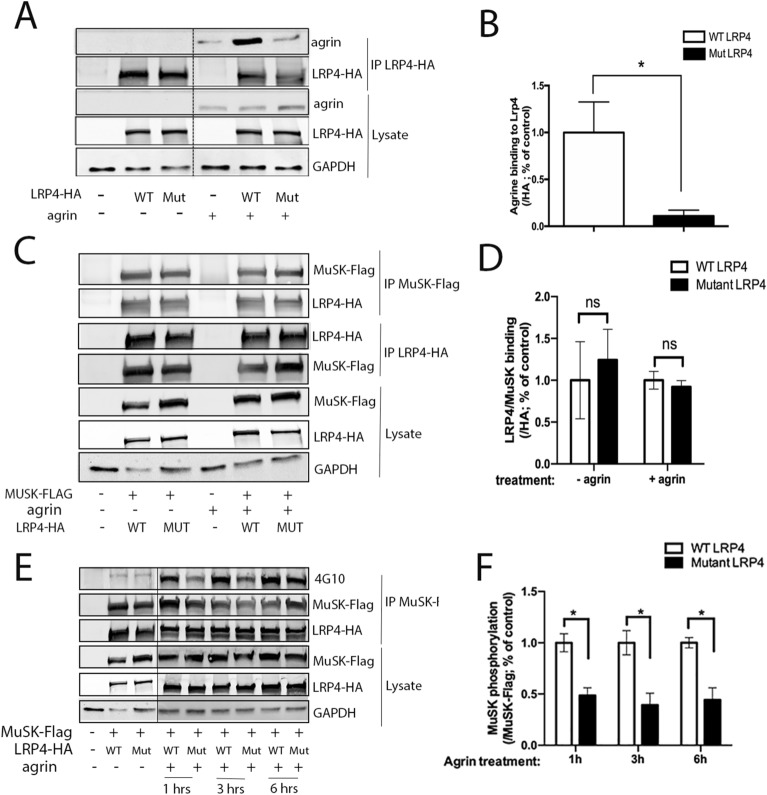
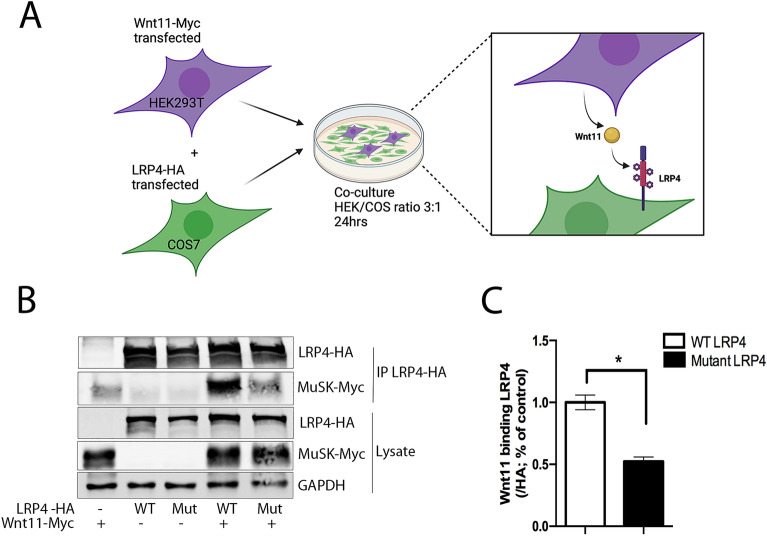
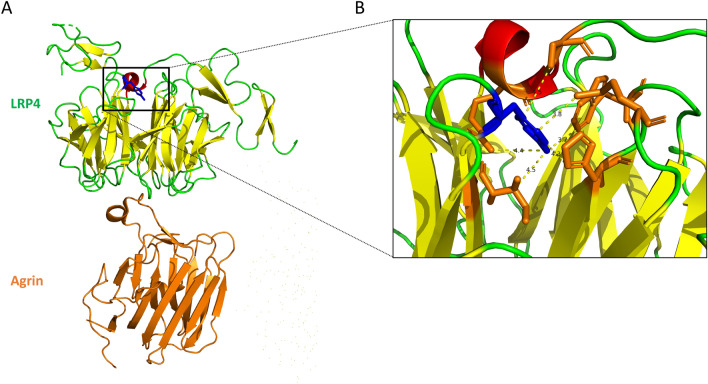
Congenital myasthenic syndromes (CMS) are a clinically and genetically heterogeneous group of rare diseases due to mutations in neuromuscular junction (NMJ) protein-coding genes. Until now, many mutations encoding postsynaptic proteins as Agrin, MuSK and LRP4 have been identified as responsible for increasingly complex CMS phenotypes. The majority of mutations identified in LRP4 gene causes bone diseases including CLS and sclerosteosis-2 and rare cases of CMS with mutations in LRP4 gene has been described so far. In the French cohort of CMS patients, we identified a novel LRP4 homozygous missense mutation (c.1820A > G; p.Thy607Cys) within the β1 propeller domain in a patient presenting CMS symptoms, including muscle weakness, fluctuating fatigability and a decrement in compound muscle action potential in spinal accessory nerves, associated with congenital agenesis of the hands and feet and renal malformation. Mechanistic expression studies show a significant decrease of AChR aggregation in cultured patient myotubes, as well as altered in vitro binding of agrin and Wnt11 ligands to the mutated β1 propeller domain of LRP4 explaining the dual phenotype characterized clinically and electoneuromyographically in the patient. These results expand the LRP4 mutations spectrum associated with a previously undescribed clinical association involving impaired neuromuscular transmission and limb deformities and highlighting the critical role of a yet poorly described domain of LRP4 at the NMJ. This study raises the question of the frequency of this rare neuromuscular form and the future diagnosis and management of these cases.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Miscellaneous
