

Ariadne: synthetic long read deconvolution using assembly graphs
- PMID: 37641111
- PMCID: PMC10463629
- DOI: 10.1186/s13059-023-03033-5
Ariadne: synthetic long read deconvolution using assembly graphs
Abstract
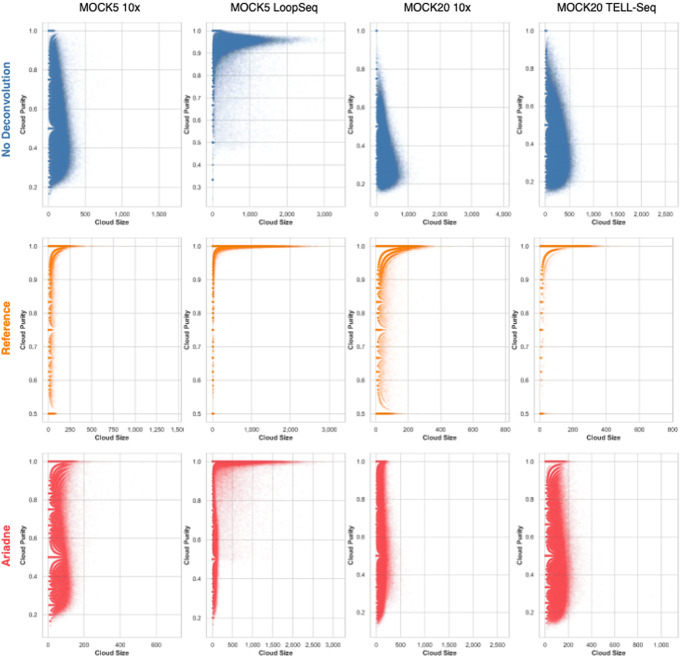
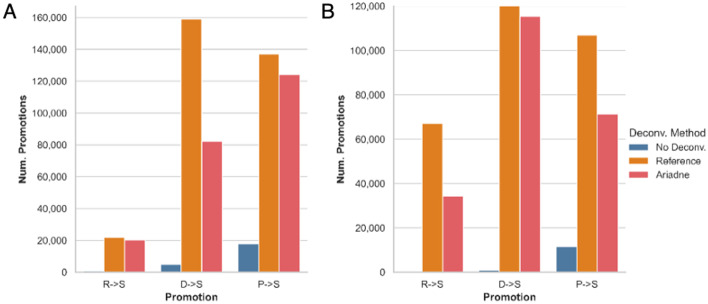
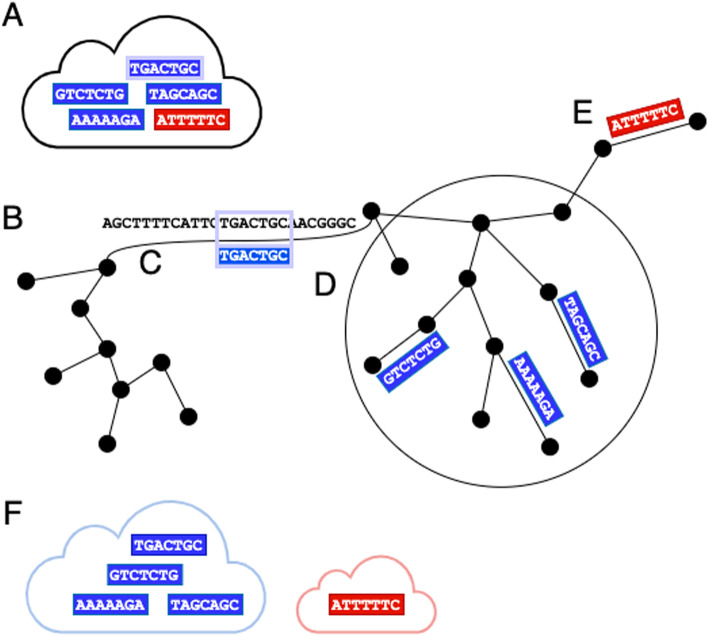
Synthetic long read sequencing techniques such as UST's TELL-Seq and Loop Genomics' LoopSeq combine 3[Formula: see text] barcoding with standard short-read sequencing to expand the range of linkage resolution from hundreds to tens of thousands of base-pairs. However, the lack of a 1:1 correspondence between a long fragment and a 3[Formula: see text] unique molecular identifier confounds the assignment of linkage between short reads. We introduce Ariadne, a novel assembly graph-based synthetic long read deconvolution algorithm, that can be used to extract single-species read-clouds from synthetic long read datasets to improve the taxonomic classification and de novo assembly of complex populations, such as metagenomes.
Keywords: Assembly graphs; Barcode deconvolution; Metagenomics; Synthetic long read.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
