

Wolman disease presenting with hemophagocytic lymphohistiocytosis syndrome and a novel LIPA gene variant: a case report and review of the literature
- PMID: 37641143
- PMCID: PMC10463876
- DOI: 10.1186/s13256-023-04116-4
Wolman disease presenting with hemophagocytic lymphohistiocytosis syndrome and a novel LIPA gene variant: a case report and review of the literature
Abstract
Background: Wolman disease is a rare disease caused by the absence of functional liposomal acid lipase due to mutations in LIPA gene. It presents with organomegaly, malabsorption, and adrenal calcifications. The presentations can resemble hemophagocytic lymphohistiocytosis, the life threatening hyperinflammatory disorder. Since the disease is very rare, clinicians might not think of it when a patient presents with hemophagocytic lymphohistiocytosis, and the opportunity to treat it properly can be lost, thus leading to demise of the child.
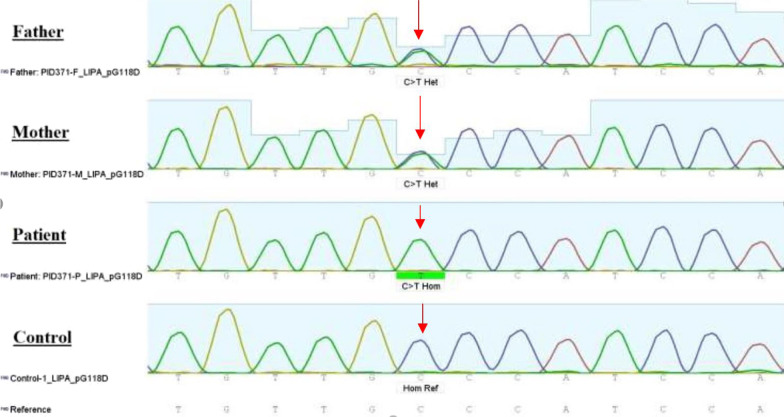
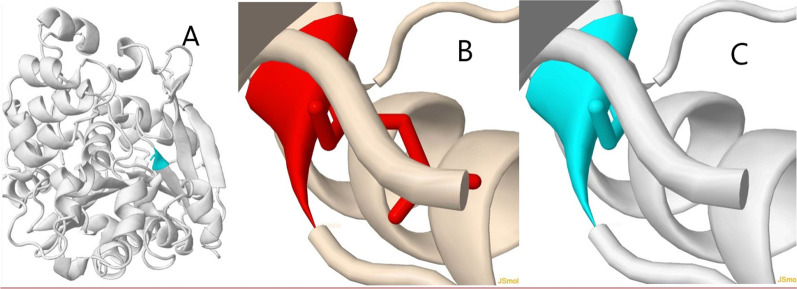
Case presentation: We present a 4.5-month-old Caucasian boy with fever, icterus, and hepatosplenomegaly who was treated according to presumed hemophagocytic lymphohistiocytosis disease. Wolman disease was diagnosed after the death of the child. There are some case reports in the literature presenting patients with Wolman disease primarily diagnosed as hemophagocytic lymphohistiocytosis, which we discuss in this review. The genetic analysis revealed after his demise was compatible with Wolman disease, introducing a novel mutation in LIPA gene: exon 4: NM_001127605: c. G353A (p.G118D), which converts the glycine amino acid to aspartic acid.
Conclusions: Considering the similarities in presentation of Wolman disease and hemophagocytic lymphohistiocytosis, the patient's life can be saved if special attention is paid to presenting features of a patient with suspected hemophagocytic lymphohistiocytosis, that is special attention to symptoms, findings on physical exams, laboratory values, and radiologic findings, and the proper treatment is urgently initiated. Reporting the novel mutations of Wolman disease can help geneticists interpret the results of their patients' genetic studies appropriately, leading to correct diagnosis and treatment.
Keywords: HLH; Hemophagocytic lymphohistiocytosis; LIPA gene; Wolman disease.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
There are no competing interests to declare.
Figures

Similar articles
-
A Novel Mutation c.153 C>A in a Tunisian Girl With Wolman Disease and Unusual Presentation: Hemophagocytic Lymphohistiocytosis.J Pediatr Hematol Oncol. 2019 Apr;41(3):e193-e196. doi: 10.1097/MPH.0000000000001192. J Pediatr Hematol Oncol. 2019. PMID: 29702543
-
Wolman's disease presenting with secondary hemophagocytic lymphohistiocytosis: a case report from Saudi Arabia and literature review.BMC Pediatr. 2021 Feb 10;21(1):72. doi: 10.1186/s12887-021-02541-2. BMC Pediatr. 2021. PMID: 33568092 Free PMC article. Review.
-
Hemophagocytic Lymphohistiocytosis: A Rare Complication of an Ultrarare Lysosomal Storage Disease.J Pediatr Hematol Oncol. 2020 May;42(4):310-312. doi: 10.1097/MPH.0000000000001552. J Pediatr Hematol Oncol. 2020. PMID: 31318819
-
Clinical insights from Wolman disease: Evaluating infantile hepatosplenomegaly.Am J Med Genet A. 2022 Nov;188(11):3364-3368. doi: 10.1002/ajmg.a.62923. Epub 2022 Aug 16. Am J Med Genet A. 2022. PMID: 35972026
-
Rare diseases presenting with hemophagocytic lymphohistiocytosis.Pediatr Int. 2023 Jan-Dec;65(1):e15516. doi: 10.1111/ped.15516. Pediatr Int. 2023. PMID: 36843347 Review.
Cited by
-
Practical Recommendations for the Diagnosis and Management of Lysosomal Acid Lipase Deficiency with a Focus on Wolman Disease.Nutrients. 2024 Dec 13;16(24):4309. doi: 10.3390/nu16244309. Nutrients. 2024. PMID: 39770929 Free PMC article. Review.
References
-
- Zhang JR, Liang XL, Jin R, Lu G. HLH-2004 protocol: diagnostic and therapeutic guidelines for childhood hemophagocytic lymphohistiocytosis. Zhongguo Dang Dai Er Ke Za Zhi. 2013;15(8):686–688. - PubMed
-
- Santos Silva E, Klaudel-Dreszler M, Bakula A, Oliva T, Sousa T, Fernandes PC, Tylki-Szymanska A, Kamenets E, Martins E, Socha P. Early onset lysosomal acid lipase deficiency presenting as secondary hemophagocytic lymphohistiocytosis: two infants treated with sebelipase alfa. Clin Res Hepatol Gastroenterol. 2018;42(5):e77–e82. doi: 10.1016/j.clinre.2018.03.012. - DOI - PubMed
-
- Trottestam H, Horne A, Aricò M, Egeler RM, Filipovich AH, Gadner H, Imashuku S, Ladisch S, Webb D, Janka G, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–4584. doi: 10.1182/blood-2011-06-356261. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
