

A novel molecular class that recruits HDAC/MECP2 complexes to PU.1 motifs reduces neuroinflammation
- PMID: 37642942
- PMCID: PMC10465325
- DOI: 10.1084/jem.20222105
A novel molecular class that recruits HDAC/MECP2 complexes to PU.1 motifs reduces neuroinflammation
Abstract
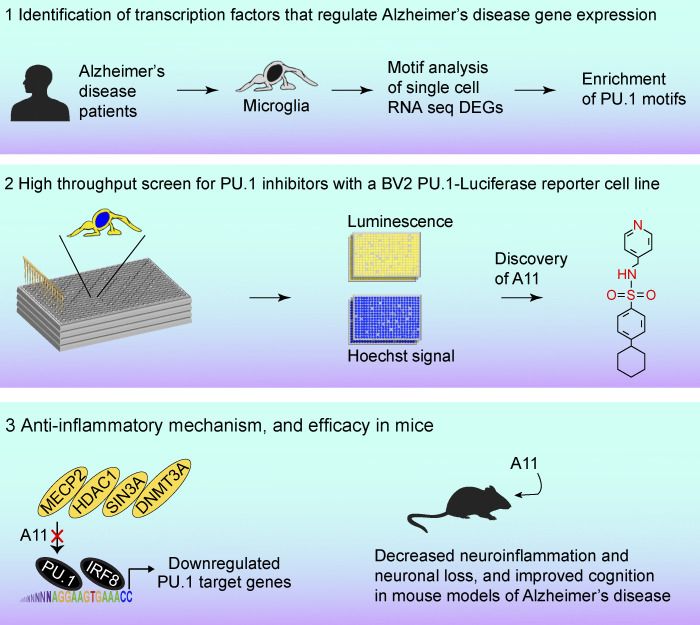
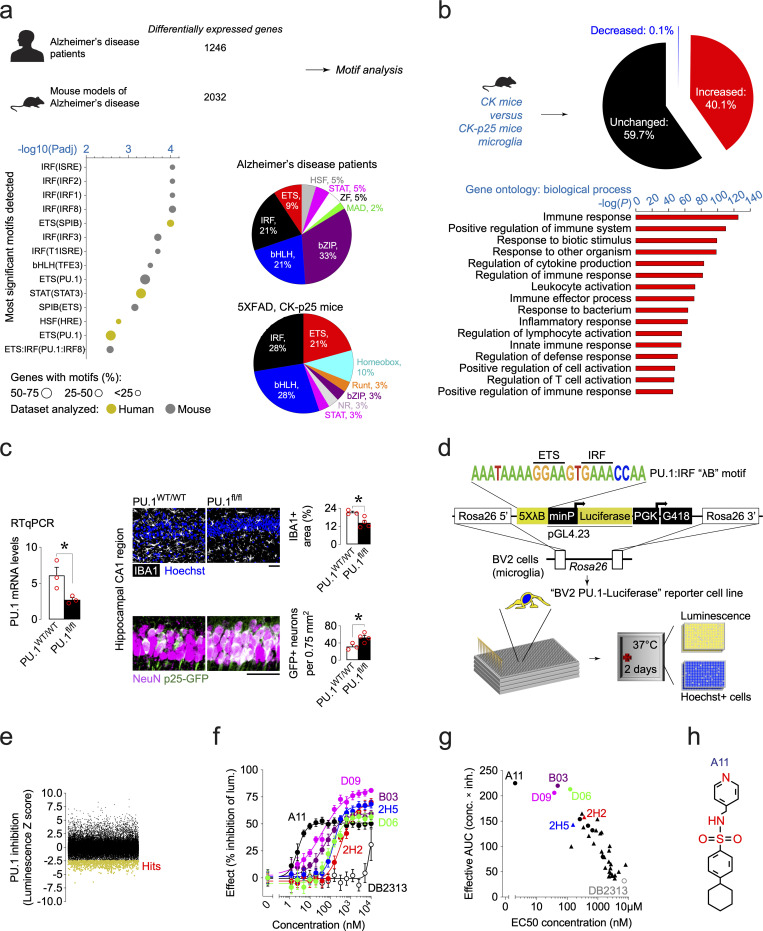
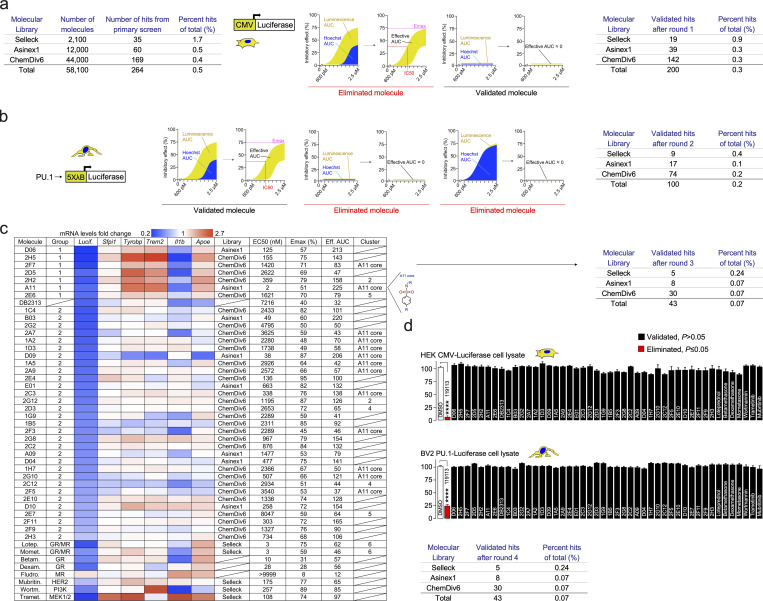
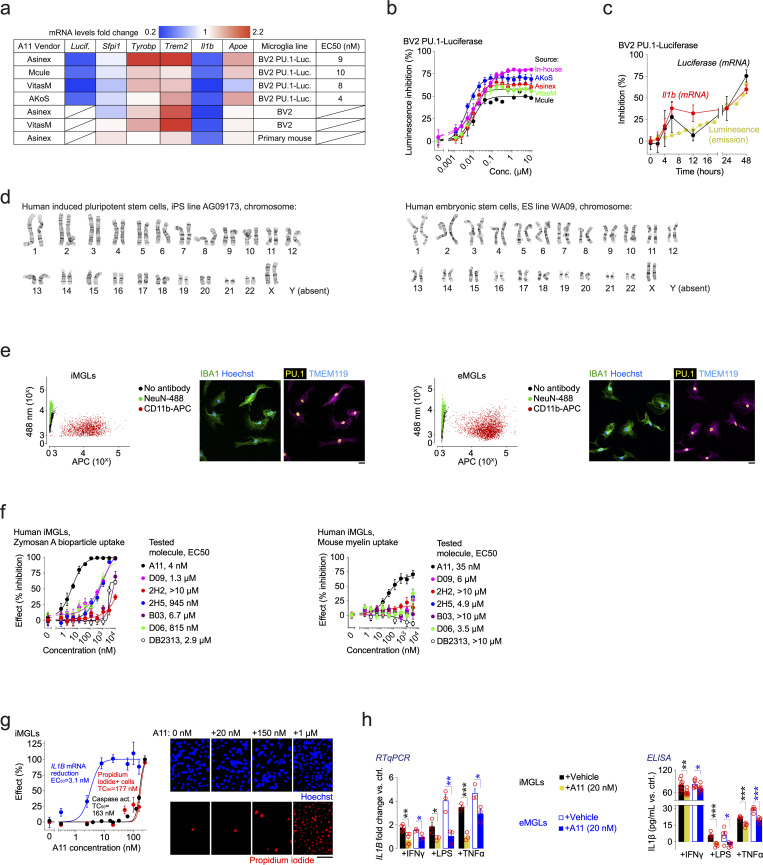
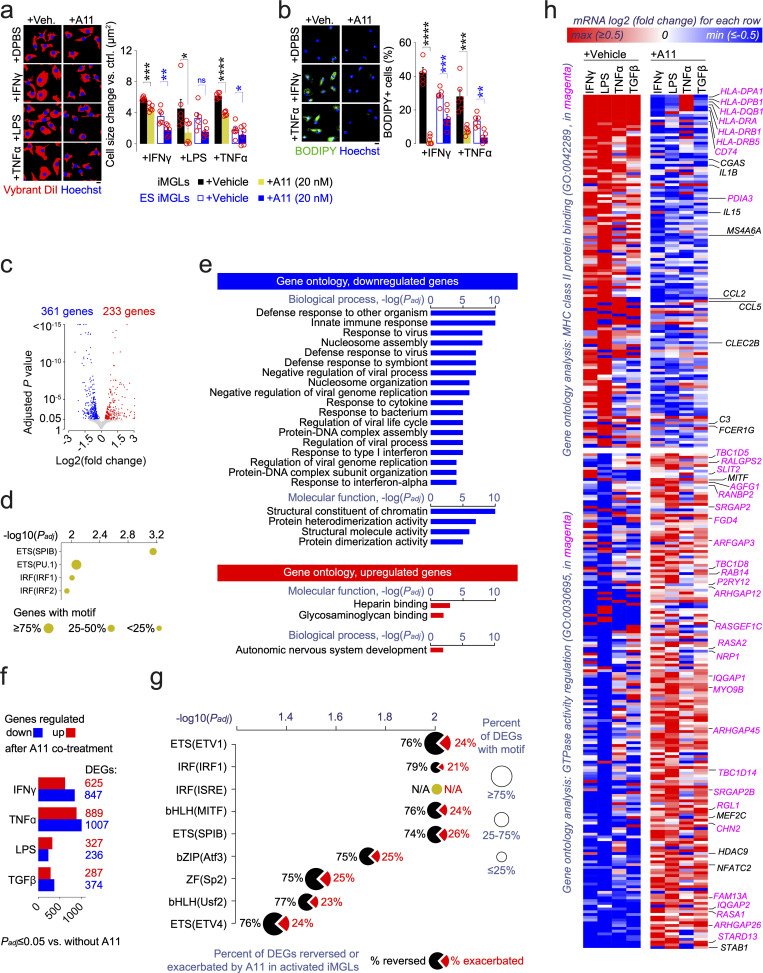
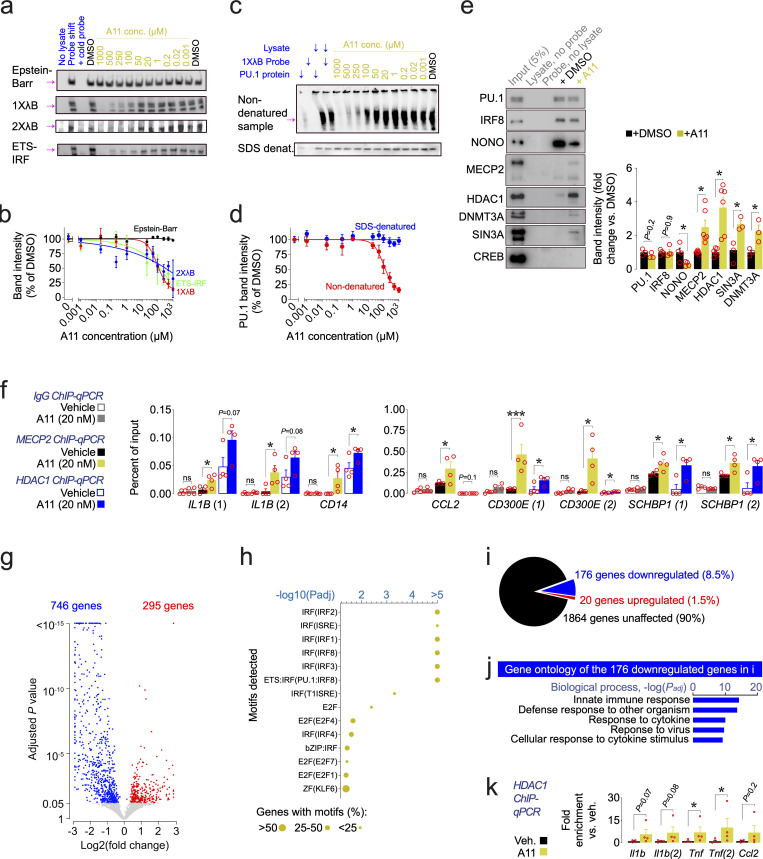
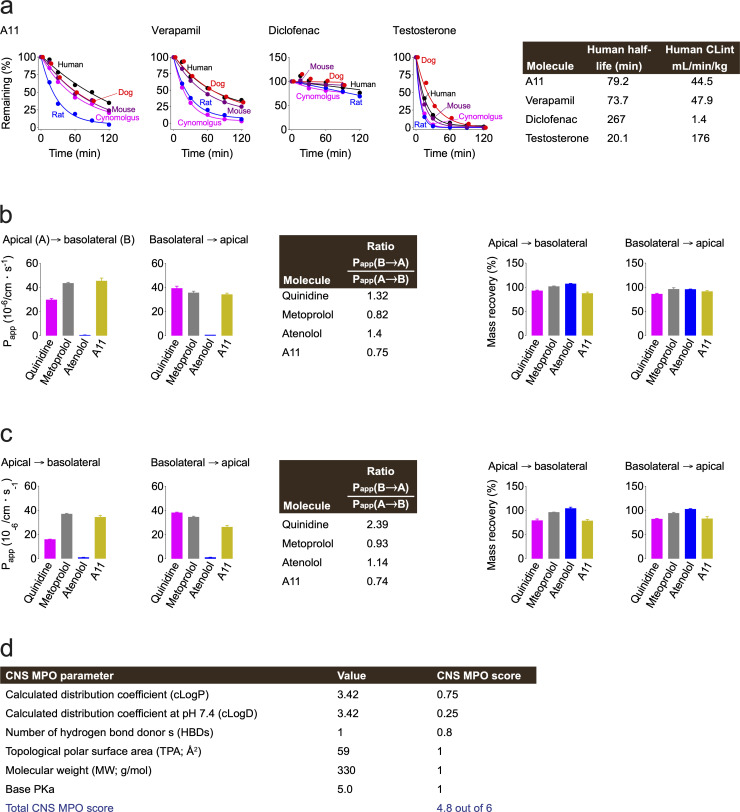
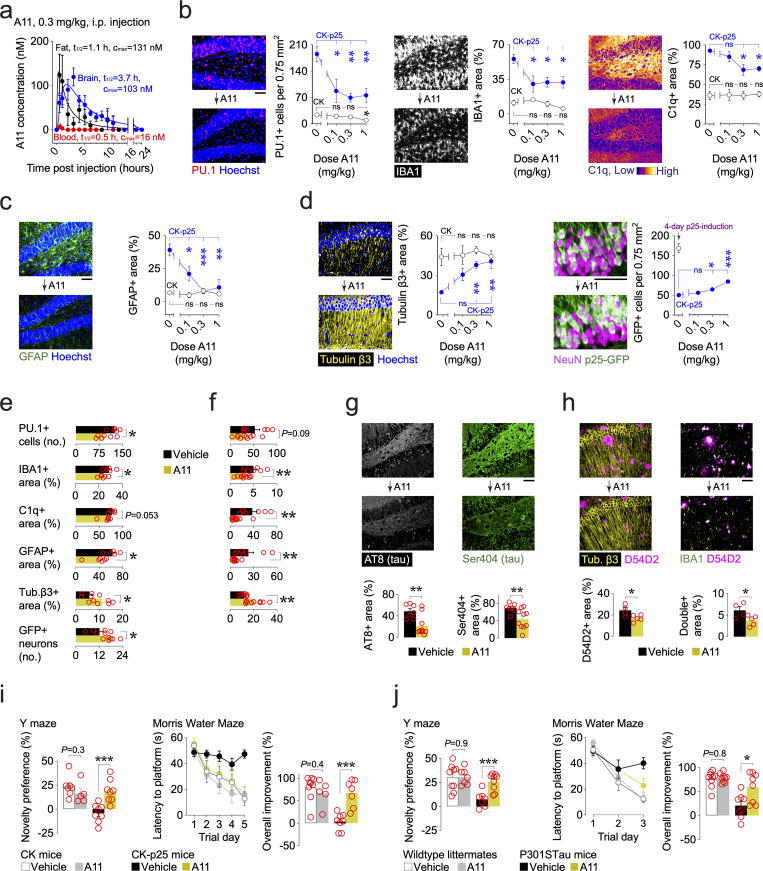
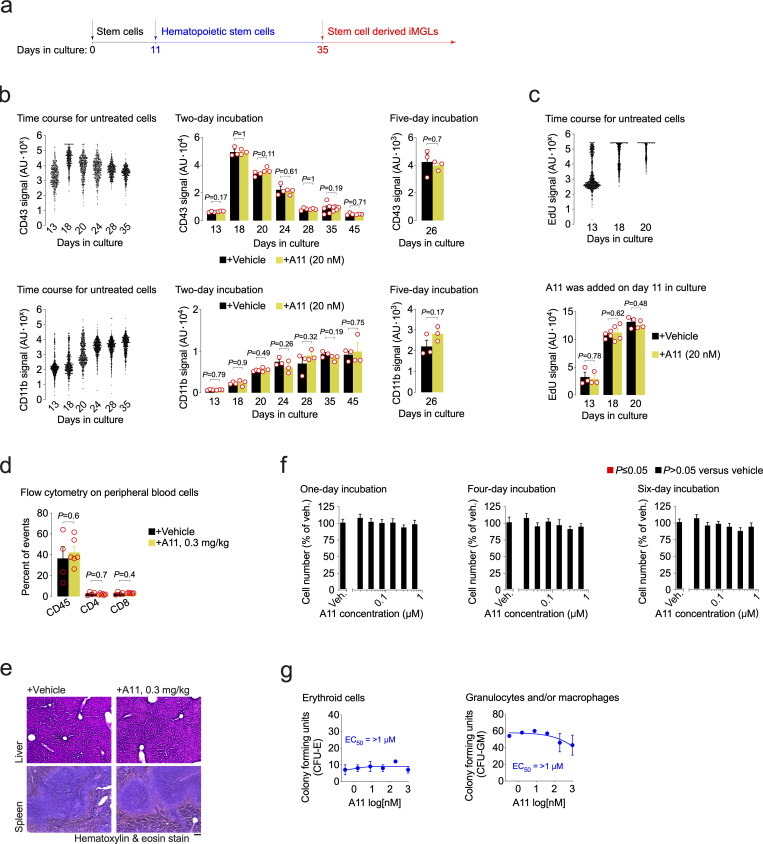
Pervasive neuroinflammation occurs in many neurodegenerative diseases, including Alzheimer's disease (AD). SPI1/PU.1 is a transcription factor located at a genome-wide significant AD-risk locus and its reduced expression is associated with delayed onset of AD. We analyzed single-cell transcriptomic datasets from microglia of human AD patients and found an enrichment of PU.1-binding motifs in the differentially expressed genes. In hippocampal tissues from transgenic mice with neurodegeneration, we found vastly increased genomic PU.1 binding. We then screened for PU.1 inhibitors using a PU.1 reporter cell line and discovered A11, a molecule with anti-inflammatory efficacy and nanomolar potency. A11 regulated genes putatively by recruiting a repressive complex containing MECP2, HDAC1, SIN3A, and DNMT3A to PU.1 motifs, thus representing a novel mechanism and class of molecules. In mouse models of AD, A11 ameliorated neuroinflammation, loss of neuronal integrity, AD pathology, and improved cognitive performance. This study uncovers a novel class of anti-inflammatory molecules with therapeutic potential for neurodegenerative disorders.
© 2023 Ralvenius et al.
Conflict of interest statement
Disclosures: W.T. Ralvenius reported patent WO2021051016A1 for systems and assays for identifying PU.1 inhibitors. C.G. Fernandez, W.J. Ray, and A. Beckmann reported a patent to WO2022217239A1 pending. A. Goate reported personal fees from Genentech, Muna Therapeutics, VIB Leuven, and Biogen outside the submitted work. L.-H. Tsai reported a patent to US-2022-0340983-A1 pending. No other disclosures were reported.
Figures

References
-
- Alzheimer, A. 1907. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Zeitschr. Psychiatr. 64:146–148.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
