

Loss of LCMT1 and biased protein phosphatase 2A heterotrimerization drive prostate cancer progression and therapy resistance
- PMID: 37644036
- PMCID: PMC10465527
- DOI: 10.1038/s41467-023-40760-6
Loss of LCMT1 and biased protein phosphatase 2A heterotrimerization drive prostate cancer progression and therapy resistance
Abstract
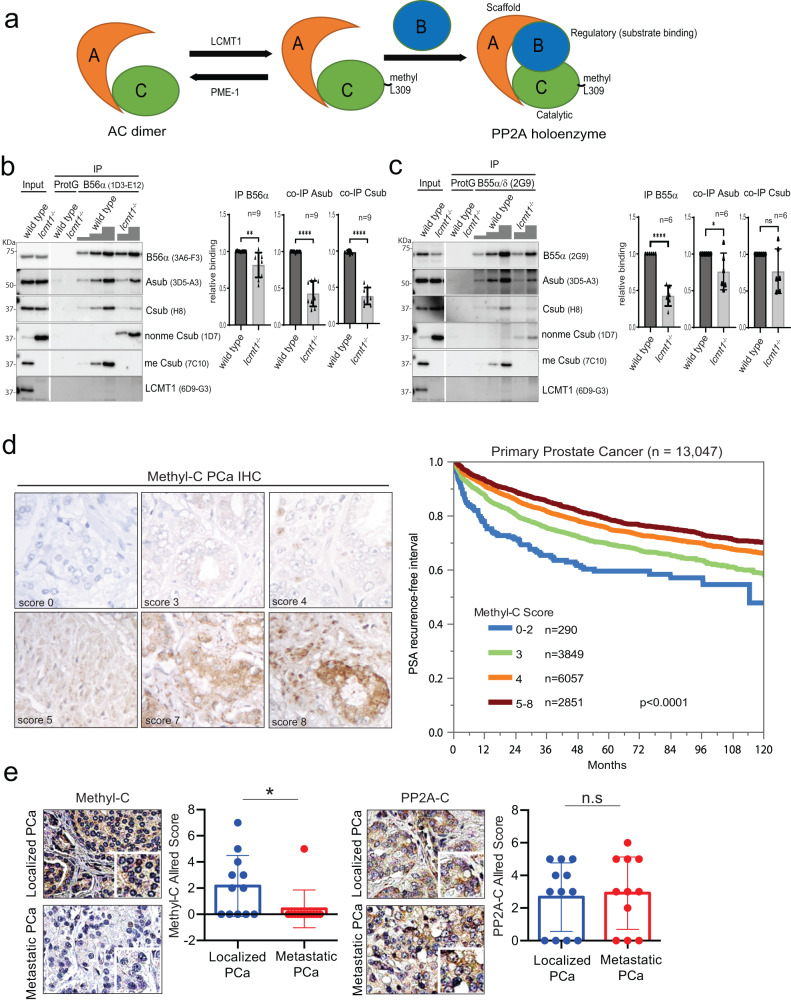
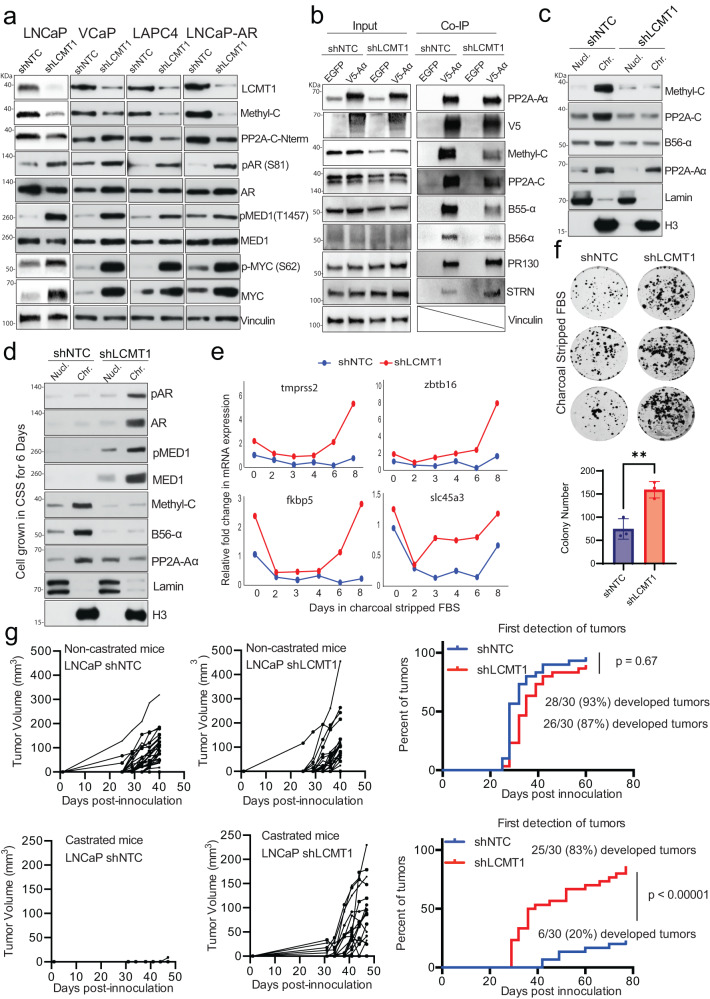
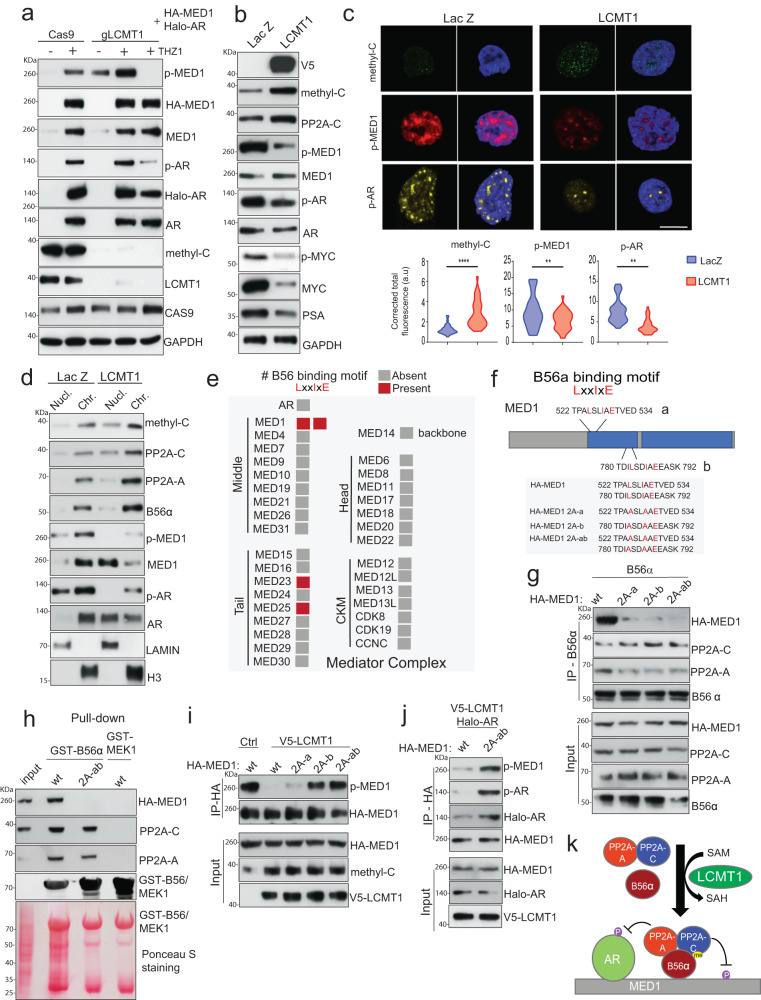
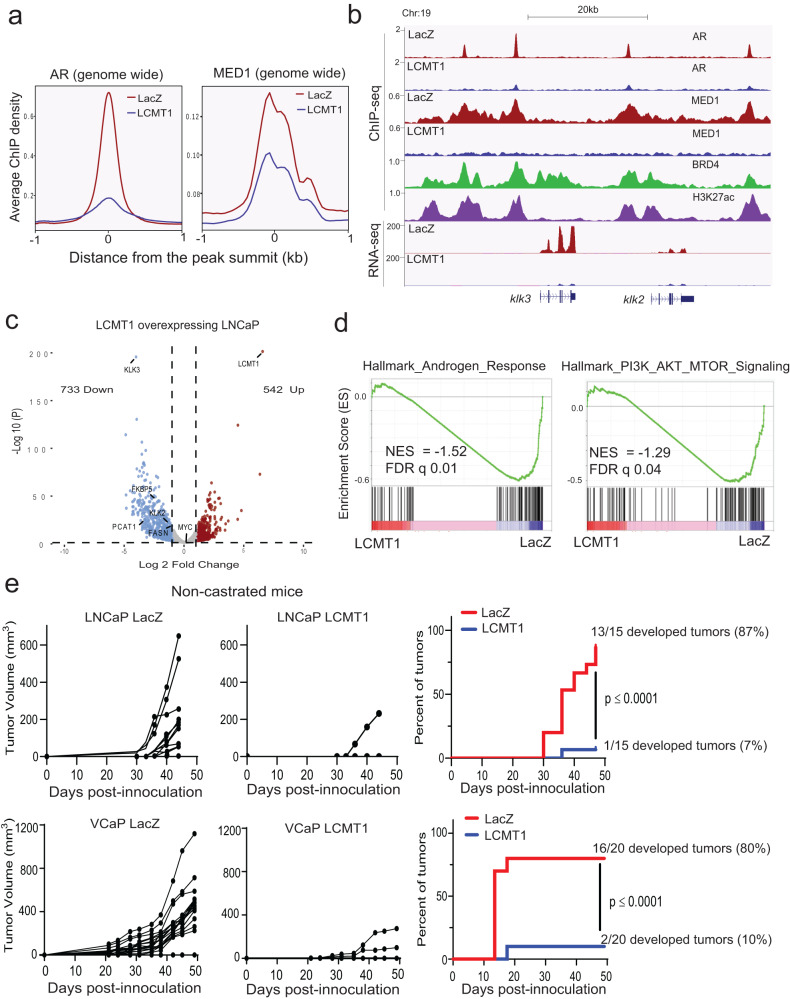
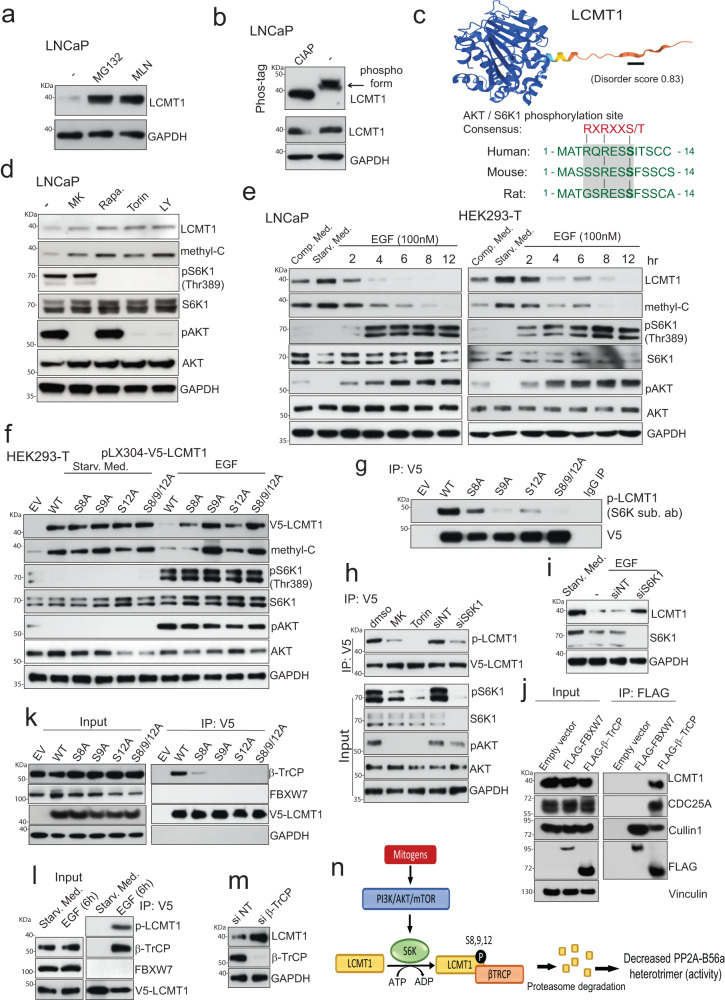
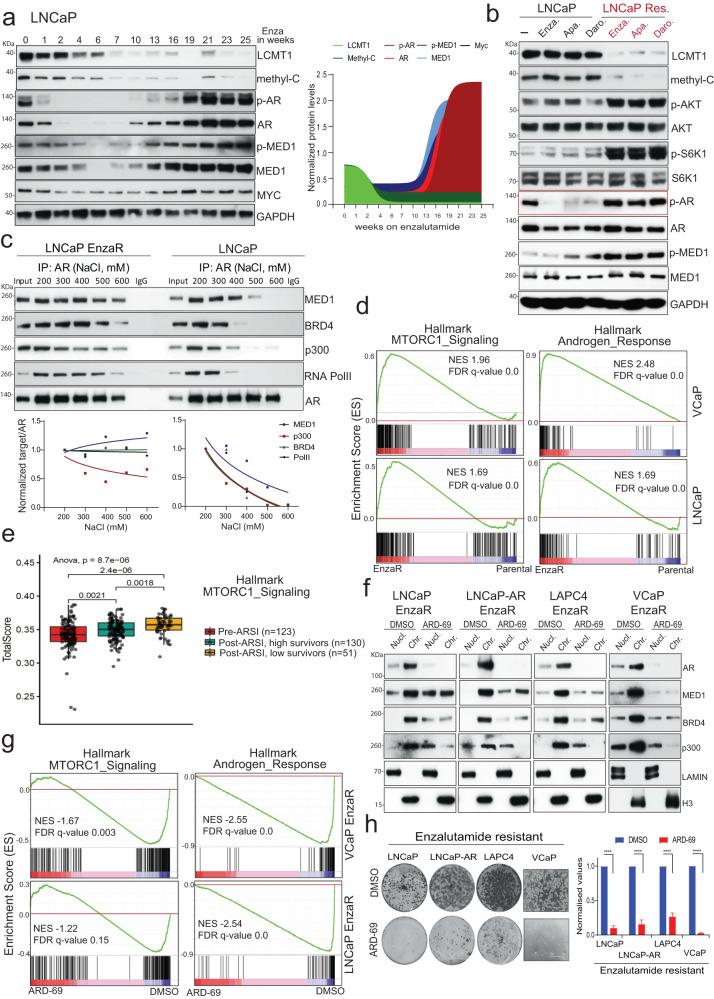
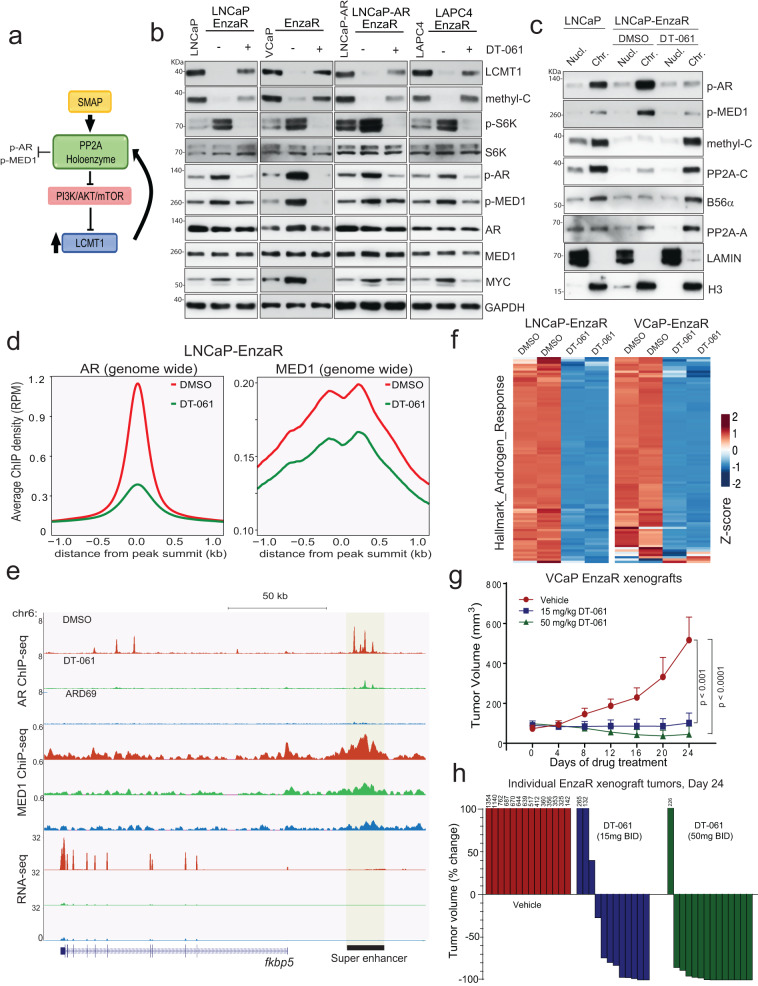
Loss of the tumor suppressive activity of the protein phosphatase 2A (PP2A) is associated with cancer, but the underlying molecular mechanisms are unclear. PP2A holoenzyme comprises a heterodimeric core, a scaffolding A subunit and a catalytic C subunit, and one of over 20 distinct substrate-directing regulatory B subunits. Methylation of the C subunit regulates PP2A heterotrimerization, affecting B subunit binding and substrate specificity. Here, we report that the leucine carboxy methyltransferase (LCMT1), which methylates the L309 residue of the C subunit, acts as a suppressor of androgen receptor (AR) addicted prostate cancer (PCa). Decreased methyl-PP2A-C levels in prostate tumors is associated with biochemical recurrence and metastasis. Silencing LCMT1 increases AR activity and promotes castration-resistant prostate cancer growth. LCMT1-dependent methyl-sensitive AB56αCme heterotrimers target AR and its critical coactivator MED1 for dephosphorylation, resulting in the eviction of the AR-MED1 complex from chromatin and loss of target gene expression. Mechanistically, LCMT1 is regulated by S6K1-mediated phosphorylation-induced degradation requiring the β-TRCP, leading to acquired resistance to anti-androgens. Finally, feedforward stabilization of LCMT1 by small molecule activator of phosphatase (SMAP) results in attenuation of AR-signaling and tumor growth inhibition in anti-androgen refractory PCa. These findings highlight methyl-PP2A-C as a prognostic marker and that the loss of LCMT1 is a major determinant in AR-addicted PCa, suggesting therapeutic potential for AR degraders or PP2A modulators in prostate cancer treatment.
© 2023. Springer Nature Limited.
Conflict of interest statement
G.N. has an equity interest in RAPPTA Therapeutics and serves as consultants, along with C.O. E.O. serves as a consultant to Millipore Corporation and RAPPTA Therapeutics and has option interests in RAPPTA Therapeutics. The Medical University of Vienna and the Regents of The University of Michigan on behalf of the authors EO and GN have filed patent WO2021148681A1 on the PP2A methyl-C subunit specific monoclonal antibody 7C10-C5 with the title: Antibodies specifically binding the carboxymethylated catalytic subunit of protein phosphatase 2A. All other authors declare no competing interests.
Figures

References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Brautigan DL, Shenolikar S. Protein serine/threonine phosphatases: keys to unlocking regulators and substrates. Annu. Rev. Biochem. 2018;87:921–964. - PubMed
-
- Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. - PubMed
-
- Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys. Acta. 2009;1795:1–15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
