

Selective enrichment of plasma cell-free messenger RNA in cancer-associated extracellular vesicles
- PMID: 37644220
- PMCID: PMC10465482
- DOI: 10.1038/s42003-023-05232-z
Selective enrichment of plasma cell-free messenger RNA in cancer-associated extracellular vesicles
Abstract
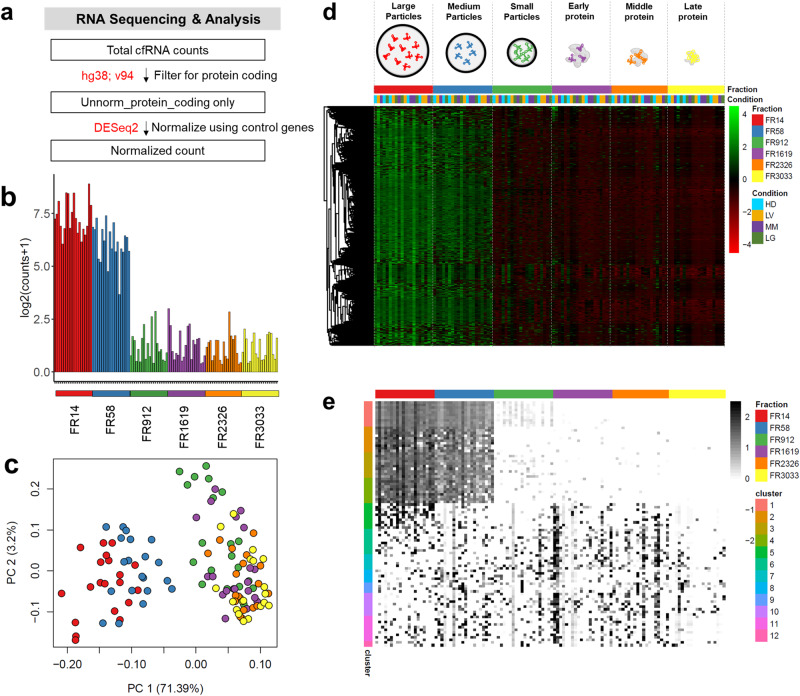
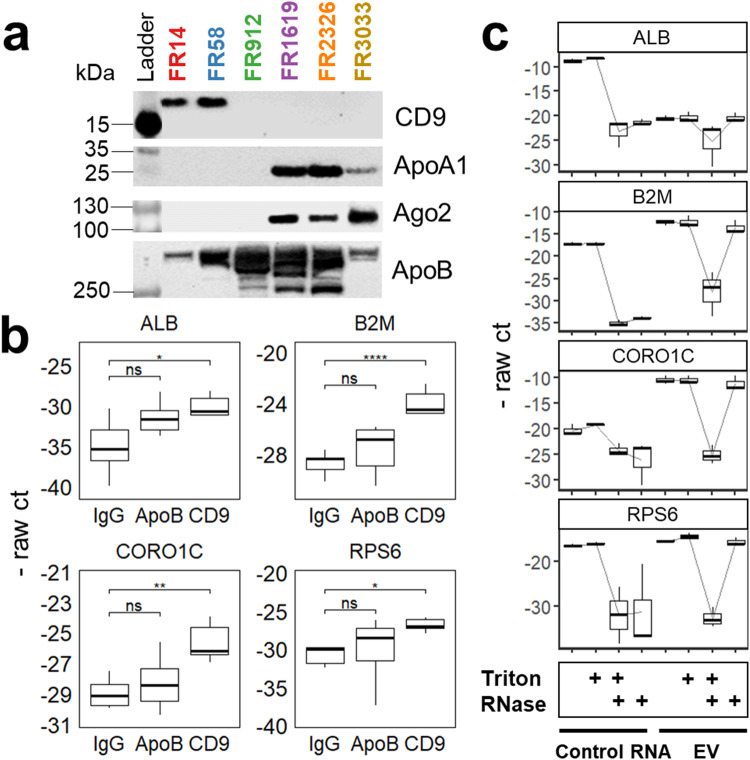
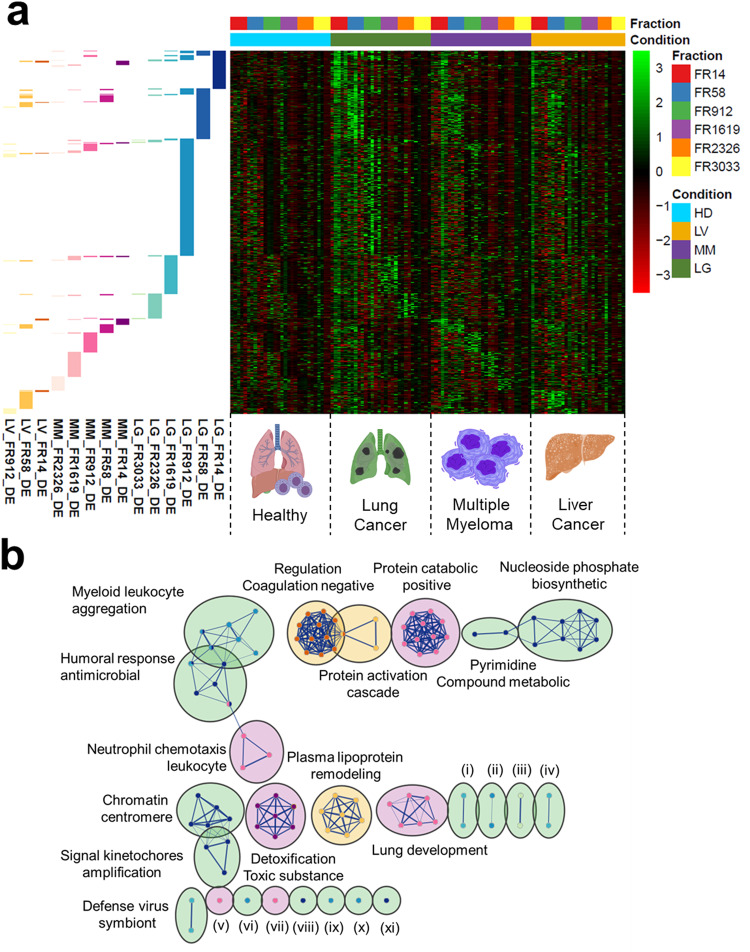
Extracellular vesicles (EVs) have been shown as key mediators of extracellular small RNA transport. However, carriers of cell-free messenger RNA (cf-mRNA) in human biofluids and their association with cancer remain poorly understood. Here, we performed a transcriptomic analysis of size-fractionated plasma from lung cancer, liver cancer, multiple myeloma, and healthy donors. Morphology and size distribution analysis showed the successful separation of large and medium particles from other soluble plasma protein fractions. We developed a strategy to purify and sequence ultra-low amounts of cf-mRNA from particle and protein enriched subpopulations with the implementation of RNA spike-ins to control for technical variability and to normalize for intrinsic drastic differences in cf-mRNA amount carried in each plasma fraction. We found that the majority of cf-mRNA was enriched and protected in EVs with remarkable stability in RNase-rich environments. We observed specific enrichment patterns of cancer-associated cf-mRNA in each particle and protein enriched subpopulation. The EV-enriched differentiating genes were associated with specific biological pathways, such as immune systems, liver function, and toxic substance regulation in lung cancer, liver cancer, and multiple myeloma, respectively. Our results suggest that dissecting the complexity of EV subpopulations illuminates their biological significance and offers a promising liquid biopsy approach.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
