

This is a preprint.
Multi-omic stratification of the missense variant cysteinome
- PMID: 37645963
- PMCID: PMC10461992
- DOI: 10.1101/2023.08.12.553095
Multi-omic stratification of the missense variant cysteinome
Update in
-
Chemoproteogenomic stratification of the missense variant cysteinome.Nat Commun. 2024 Oct 28;15(1):9284. doi: 10.1038/s41467-024-53520-x. Nat Commun. 2024. PMID: 39468056 Free PMC article.
Abstract
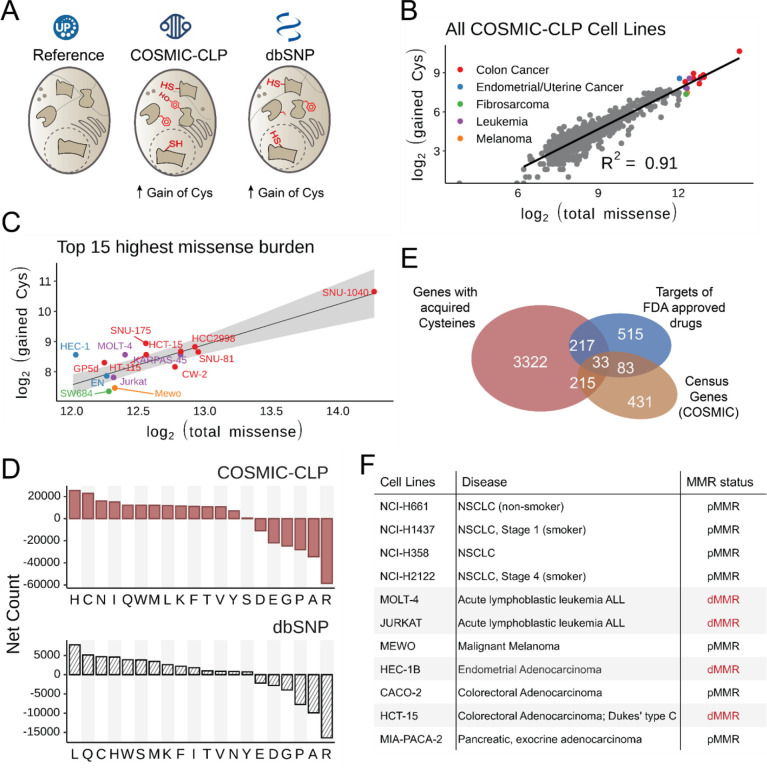
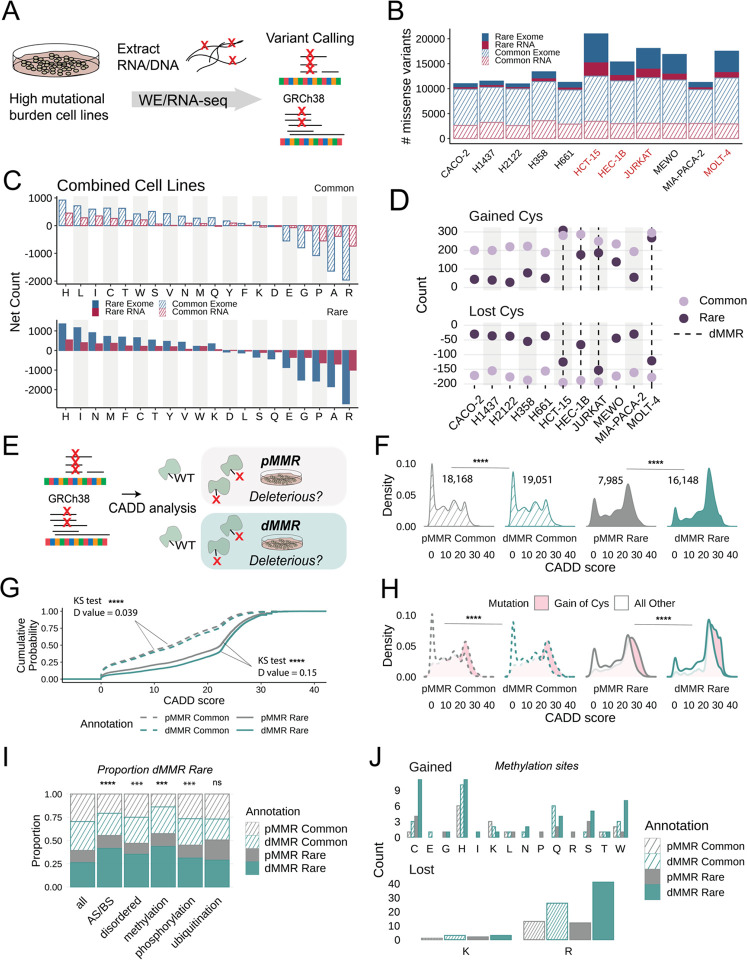
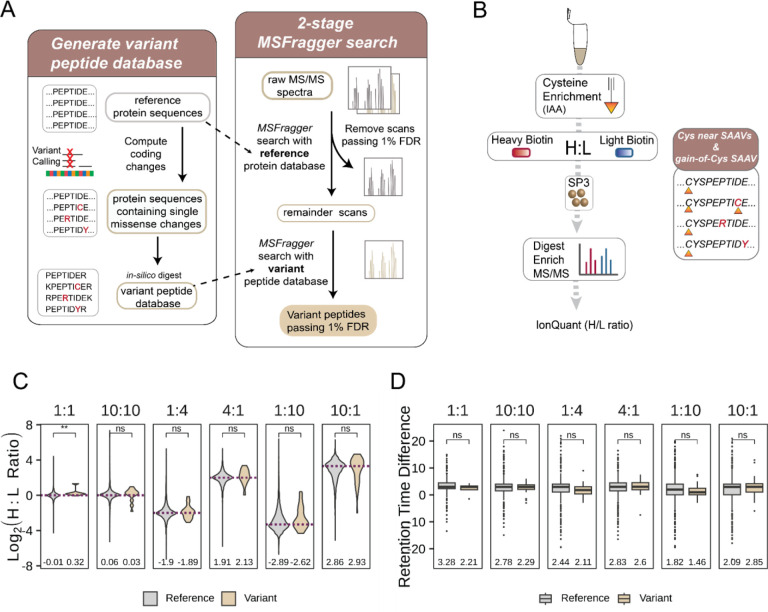
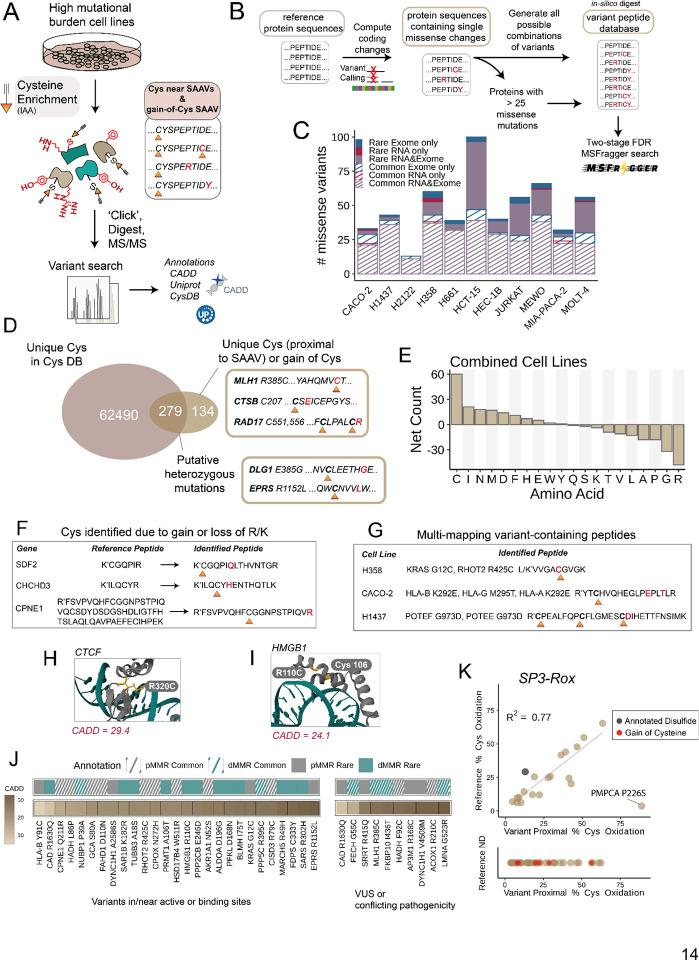
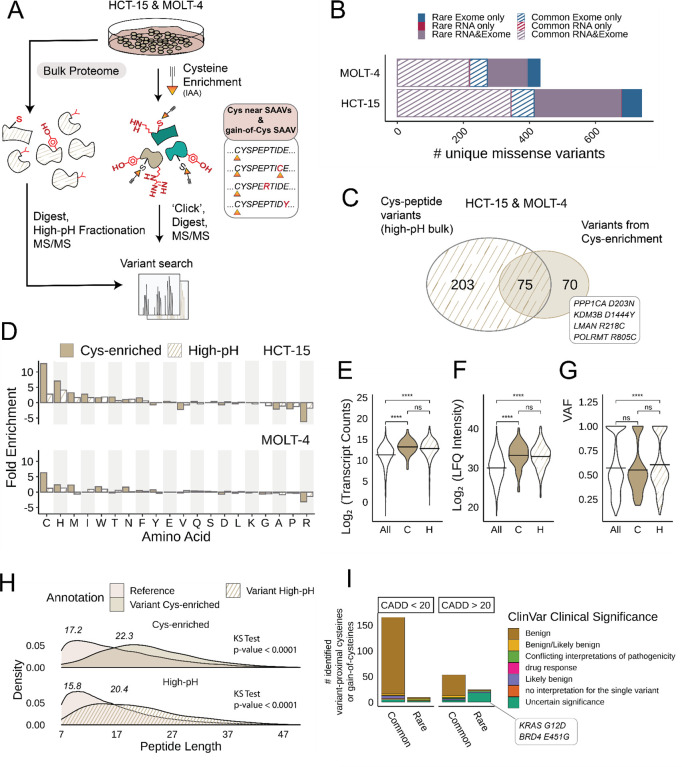
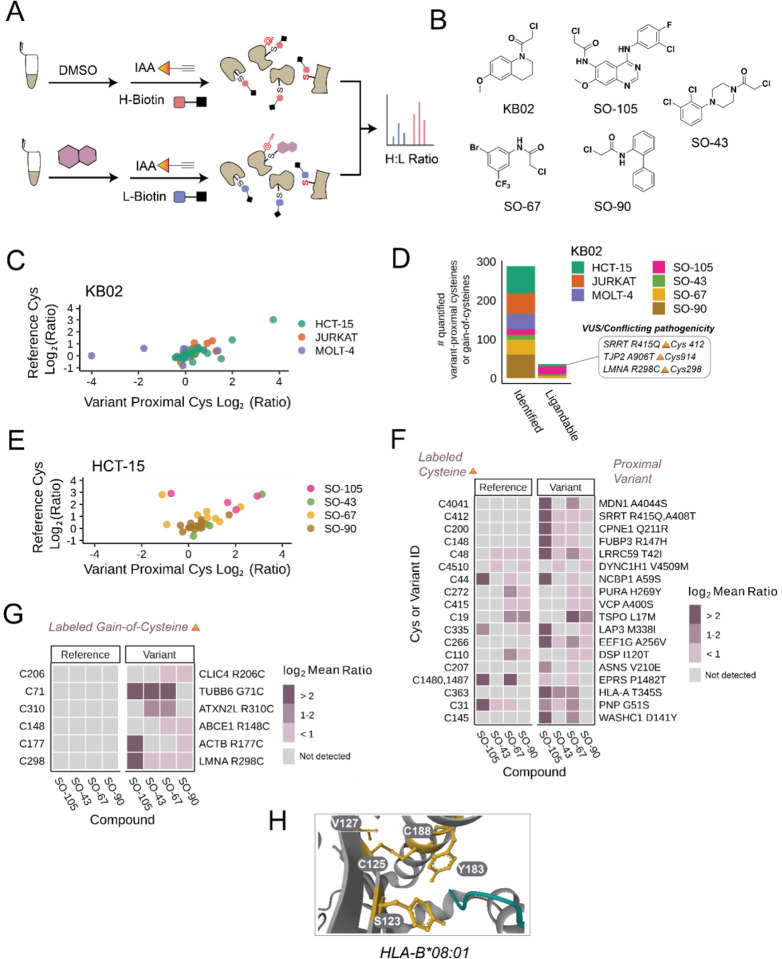
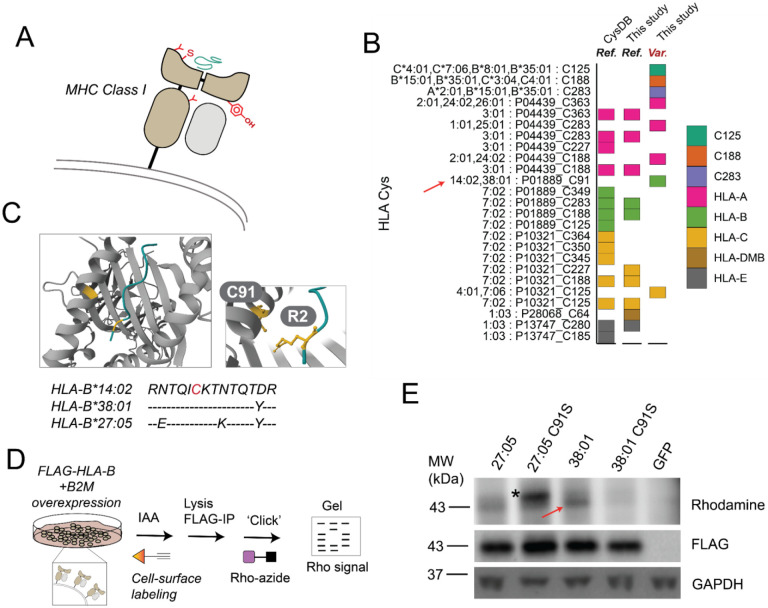
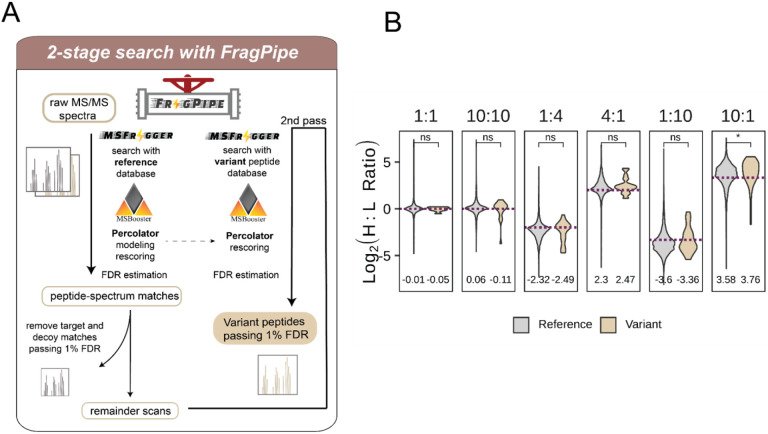
Cancer genomes are rife with genetic variants; one key outcome of this variation is gain-ofcysteine, which is the most frequently acquired amino acid due to missense variants in COSMIC. Acquired cysteines are both driver mutations and sites targeted by precision therapies. However, despite their ubiquity, nearly all acquired cysteines remain uncharacterized. Here, we pair cysteine chemoproteomics-a technique that enables proteome-wide pinpointing of functional, redox sensitive, and potentially druggable residues-with genomics to reveal the hidden landscape of cysteine acquisition. For both cancer and healthy genomes, we find that cysteine acquisition is a ubiquitous consequence of genetic variation that is further elevated in the context of decreased DNA repair. Our chemoproteogenomics platform integrates chemoproteomic, whole exome, and RNA-seq data, with a customized 2-stage false discovery rate (FDR) error controlled proteomic search, further enhanced with a user-friendly FragPipe interface. Integration of CADD predictions of deleteriousness revealed marked enrichment for likely damaging variants that result in acquisition of cysteine. By deploying chemoproteogenomics across eleven cell lines, we identify 116 gain-of-cysteines, of which 10 were liganded by electrophilic druglike molecules. Reference cysteines proximal to missense variants were also found to be pervasive, 791 in total, supporting heretofore untapped opportunities for proteoform-specific chemical probe development campaigns. As chemoproteogenomics is further distinguished by sample-matched combinatorial variant databases and compatible with redox proteomics and small molecule screening, we expect widespread utility in guiding proteoform-specific biology and therapeutic discovery.
Conflict of interest statement
Conflicts of Interest The authors declare no financial or commercial conflict of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials