

Characterization of molecular mechanisms driving Merkel cell polyomavirus oncogene transcription and tumorigenic potential
- PMID: 37647312
- PMCID: PMC10468096
- DOI: 10.1371/journal.ppat.1011598
Characterization of molecular mechanisms driving Merkel cell polyomavirus oncogene transcription and tumorigenic potential
Abstract
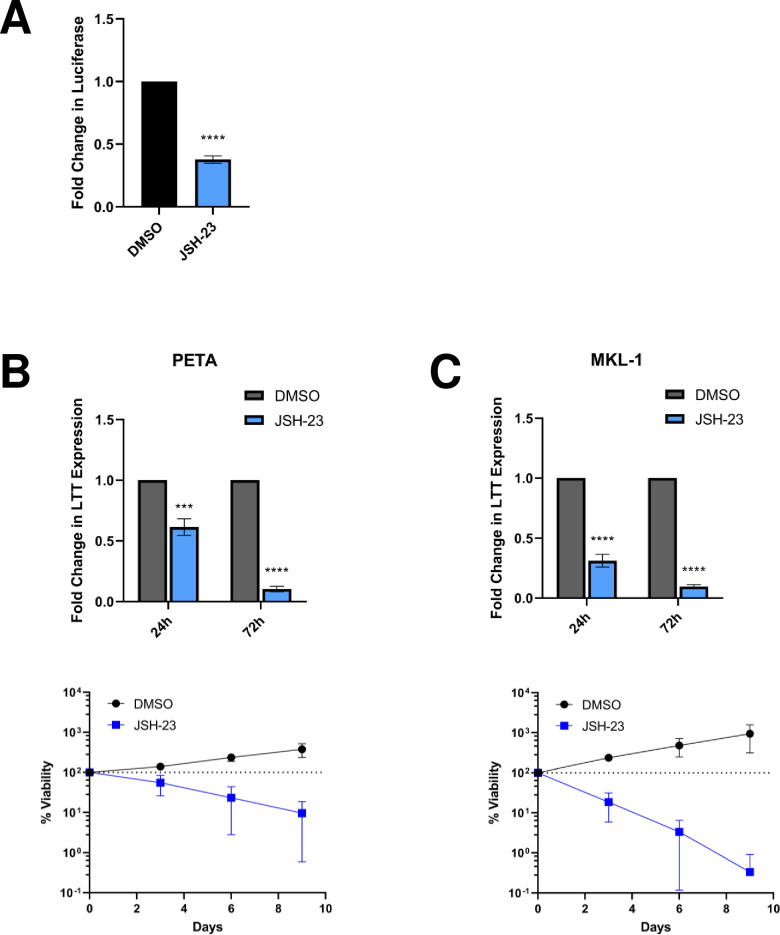
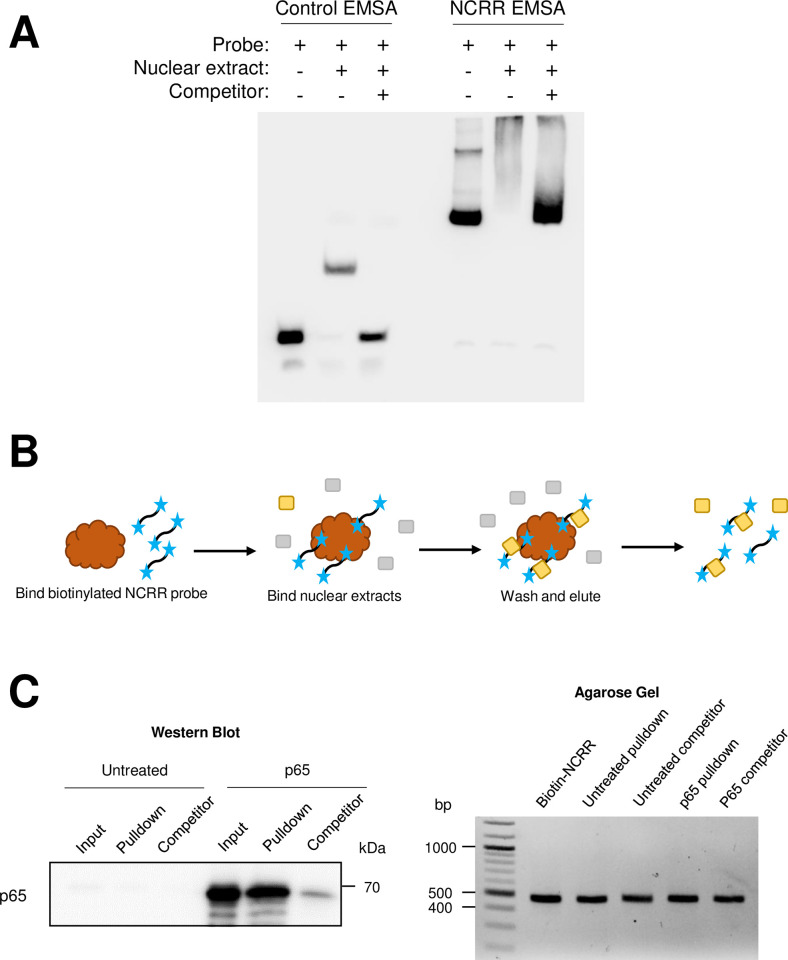
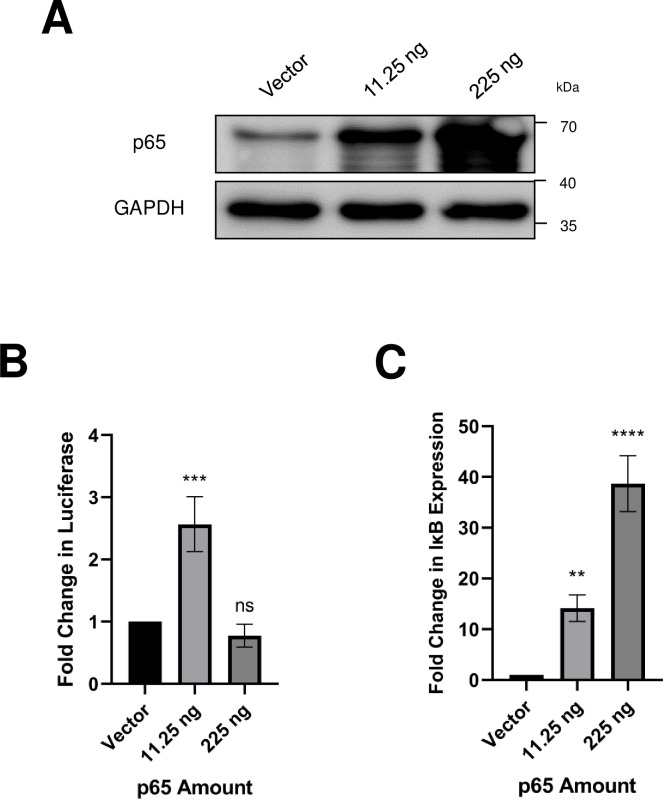
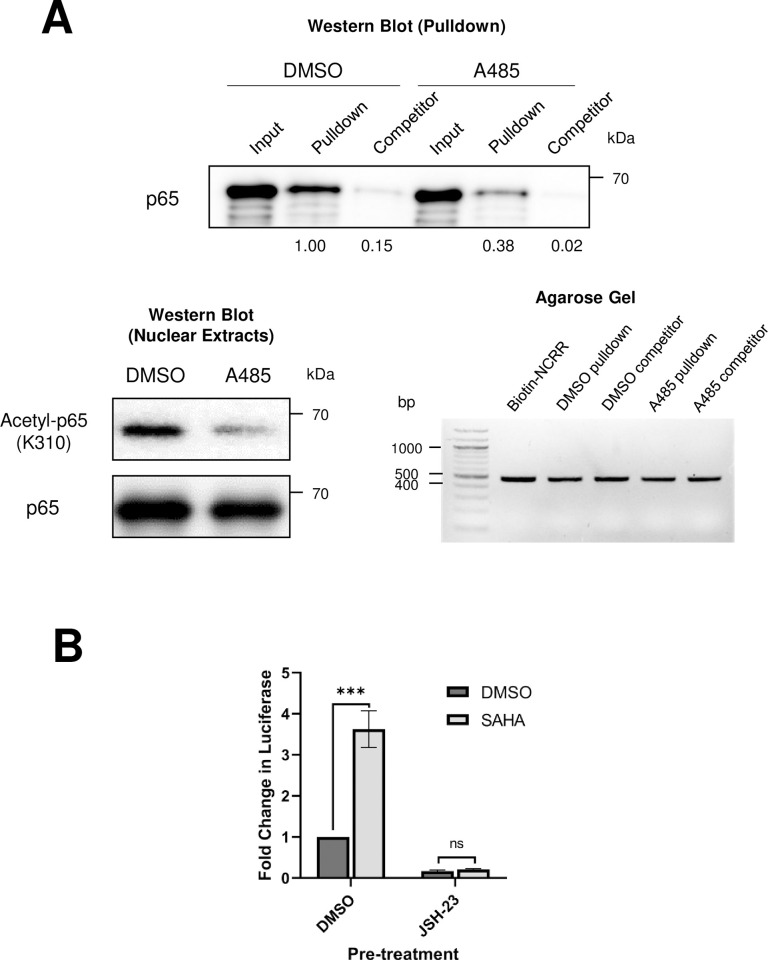
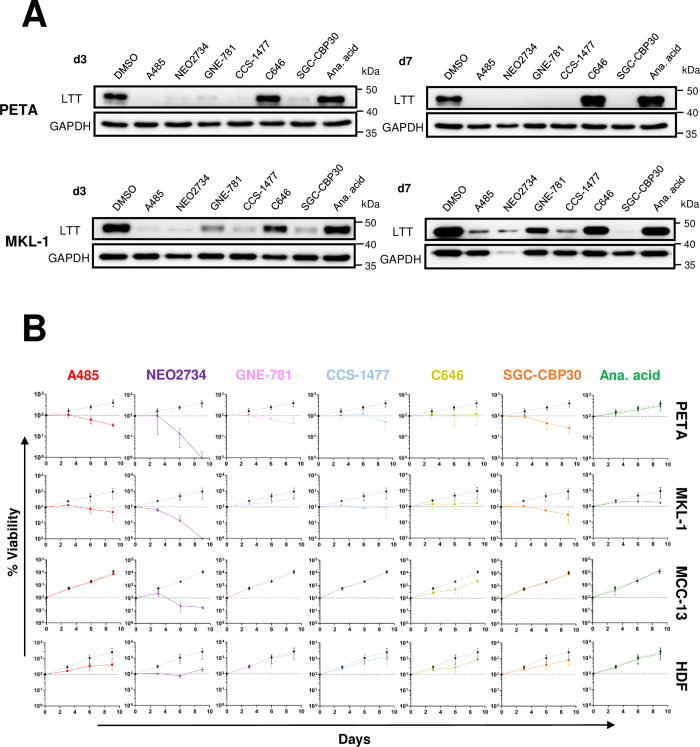
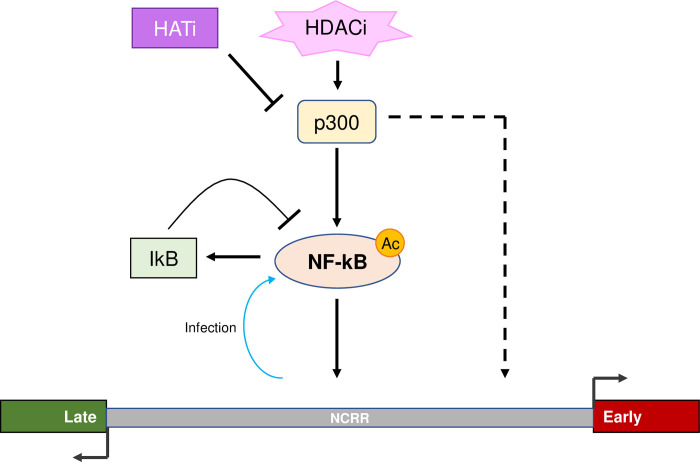
Merkel cell polyomavirus (MCPyV) is associated with approximately 80% of cases of Merkel cell carcinoma (MCC), an aggressive type of skin cancer. The incidence of MCC has tripled over the past twenty years, but there are currently very few effective targeted treatments. A better understanding of the MCPyV life cycle and its oncogenic mechanisms is needed to unveil novel strategies for the prevention and treatment of MCC. MCPyV infection and oncogenesis are reliant on the expression of the early viral oncoproteins, which drive the viral life cycle and MCPyV+ MCC tumor cell growth. To date, the molecular mechanisms regulating the transcription of the MCPyV oncogenes remain largely uncharacterized. In this study, we investigated how MCPyV early transcription is regulated to support viral infection and MCC tumorigenesis. Our studies established the roles of multiple cellular factors in the control of MCPyV gene expression. Inhibitor screening experiments revealed that the histone acetyltransferases p300 and CBP positively regulate MCPyV transcription. Their regulation of viral gene expression occurs through coactivation of the transcription factor NF-κB, which binds to the viral genome to drive MCPyV oncogene expression in a manner that is tightly controlled through a negative feedback loop. Furthermore, we discovered that small molecule inhibitors specifically targeting p300/CBP histone acetyltransferase activity are effective at blocking MCPyV tumor antigen expression and MCPyV+ MCC cell proliferation. Together, our work establishes key cellular factors regulating MCPyV transcription, providing the basis for understanding the largely unknown mechanisms governing MCPyV transcription that defines its infectious host cell tropism, viral life cycle, and oncogenic potential. Our studies also identify a novel therapeutic strategy against MCPyV+ MCC through specific blockage of MCPyV oncogene expression and MCC tumor growth.
Copyright: © 2023 Yang et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
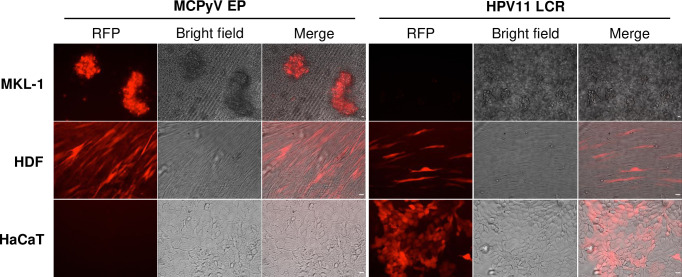
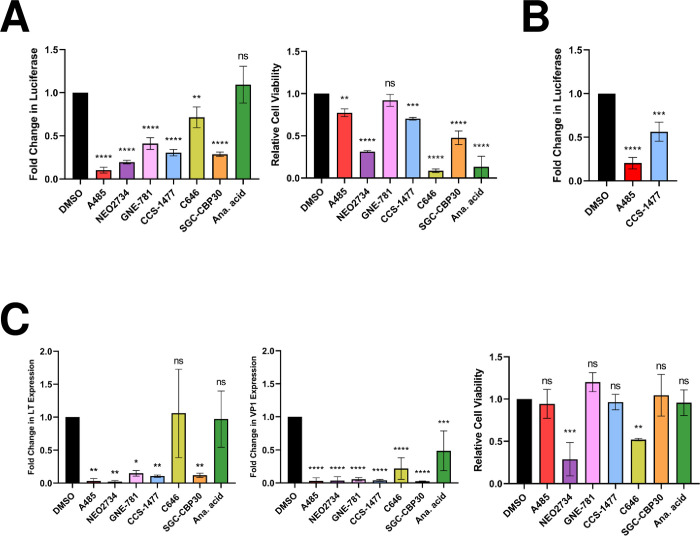
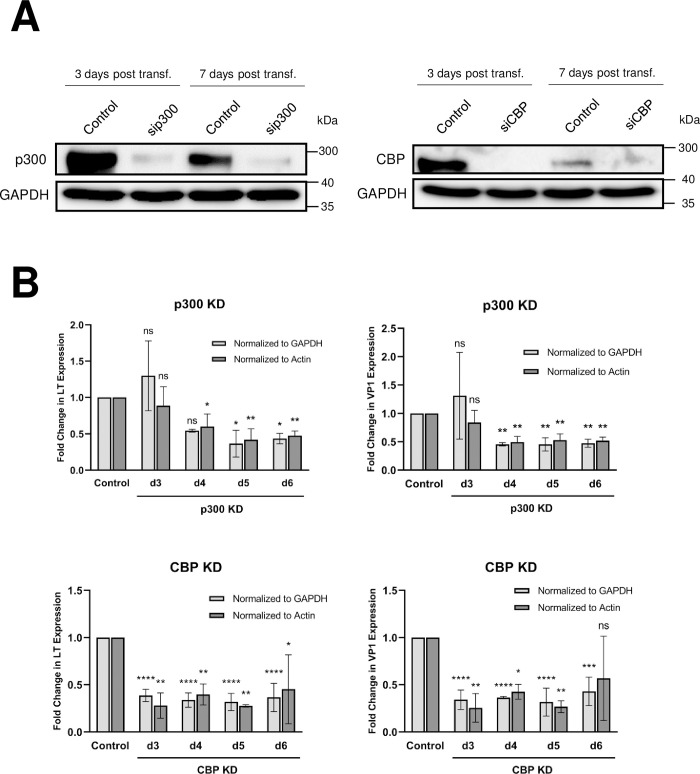
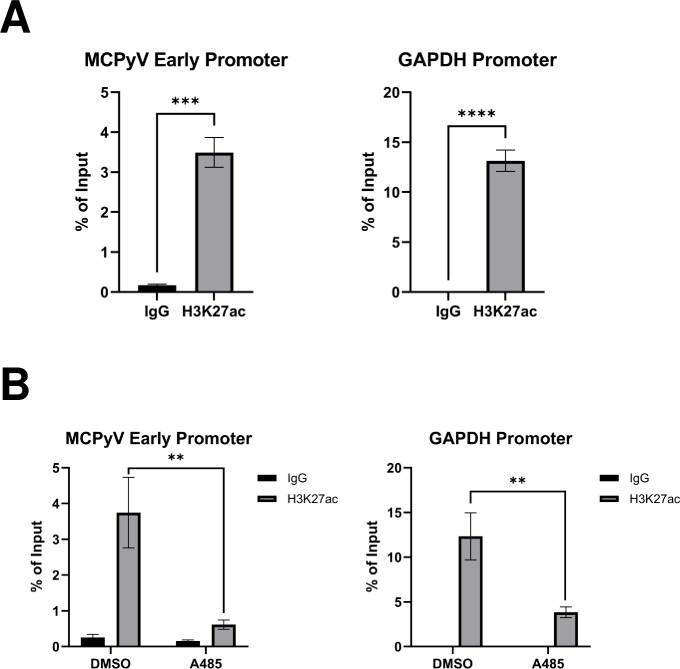
Similar articles
-
Merkel Cell Polyomavirus Downregulates N-myc Downstream-Regulated Gene 1, Leading to Cellular Proliferation and Migration.J Virol. 2020 Jan 17;94(3):e00899-19. doi: 10.1128/JVI.00899-19. Print 2020 Jan 17. J Virol. 2020. PMID: 31694959 Free PMC article.
-
Merkel Cell Polyomavirus Infection Induces an Antiviral Innate Immune Response in Human Dermal Fibroblasts.J Virol. 2021 Jun 10;95(13):e0221120. doi: 10.1128/JVI.02211-20. Epub 2021 Jun 10. J Virol. 2021. PMID: 33883226 Free PMC article.
-
Merkel cell polyomavirus and associated Merkel cell carcinoma.Tumour Virus Res. 2022 Jun;13:200232. doi: 10.1016/j.tvr.2021.200232. Epub 2021 Dec 15. Tumour Virus Res. 2022. PMID: 34920178 Free PMC article. Review.
-
Merkel Cell Polyomavirus: A New DNA Virus Associated with Human Cancer.Adv Exp Med Biol. 2017;1018:35-56. doi: 10.1007/978-981-10-5765-6_4. Adv Exp Med Biol. 2017. PMID: 29052131 Review.
-
Characterization of the Impact of Merkel Cell Polyomavirus-Induced Interferon Signaling on Viral Infection.J Virol. 2023 Apr 27;97(4):e0190722. doi: 10.1128/jvi.01907-22. Epub 2023 Mar 22. J Virol. 2023. PMID: 36946735 Free PMC article.
Cited by
-
Cellular Transformation by Human Cytomegalovirus.Cancers (Basel). 2024 May 22;16(11):1970. doi: 10.3390/cancers16111970. Cancers (Basel). 2024. PMID: 38893091 Free PMC article.
-
Merkel Cell Carcinoma Mimicking a Pyogenic Granuloma on the Thumb.Cureus. 2023 Dec 21;15(12):e50878. doi: 10.7759/cureus.50878. eCollection 2023 Dec. Cureus. 2023. PMID: 38249166 Free PMC article.
-
Polyomavirus ALTOs, but not MTs, downregulate viral early gene expression by activating the NF-κB pathway.bioRxiv [Preprint]. 2024 May 25:2024.05.24.595774. doi: 10.1101/2024.05.24.595774. bioRxiv. 2024. Update in: Proc Natl Acad Sci U S A. 2024 Aug 20;121(34):e2403133121. doi: 10.1073/pnas.2403133121. PMID: 38826197 Free PMC article. Updated. Preprint.
-
Polyomavirus ALTOs, but not MTs, downregulate viral early gene expression by activating the NF-κB pathway.Proc Natl Acad Sci U S A. 2024 Aug 20;121(34):e2403133121. doi: 10.1073/pnas.2403133121. Epub 2024 Aug 14. Proc Natl Acad Sci U S A. 2024. PMID: 39141346 Free PMC article.
-
Understanding Merkel Cell Carcinoma: Pathogenic Signaling, Extracellular Matrix Dynamics, and Novel Treatment Approaches.Cancers (Basel). 2025 Apr 2;17(7):1212. doi: 10.3390/cancers17071212. Cancers (Basel). 2025. PMID: 40227764 Free PMC article. Review.
References
-
- Paulson KG, Park SY, Vandeven NA, Lachance K, Thomas H, Chapuis AG, et al.. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. Journal of the American Academy of Dermatology. 2018;78(3):457–63.e2. Epub 20171102. doi: 10.1016/j.jaad.2017.10.028 ; PubMed Central PMCID: PMC5815902. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
