

Asprosin contributes to nonalcoholic fatty liver disease through regulating lipid accumulation and inflammatory response via AMPK signaling
- PMID: 37647445
- PMCID: PMC10436697
- DOI: 10.1002/iid3.947
Asprosin contributes to nonalcoholic fatty liver disease through regulating lipid accumulation and inflammatory response via AMPK signaling
Abstract
Background: Nonalcoholic fatty liver disease (NAFLD) is a primary contributor to liver-related morbidity and mortality. Asprosin has been reported to be implicated in NAFLD.
Aims: This work is to illuminate the effects of Asprosin on NAFLD and the possible downstream mechanism.
Materials & methods: The weight of NAFLD mice induced by a high-fat diet was detected. Quantitative reverse-transcription polymerase chain reaction (RT-qPCR) examined serum Asprosin expression. RT-qPCR and western blot analysis examined Asprosin expression in mice liver tissues. Intraperitoneal glucose tolerance test (IPGTT) and intraperitoneal insulin tolerance test (IPITT) were implemented. Biochemical kits tested liver enzyme levels in mice serum and liver tissues. Hematoxylin and eosin staining evaluated liver histology. Liver weight was also tested and oil red O staining estimated lipid accumulation. RT-qPCR and western blot analysis analyzed the expression of gluconeogenesis-, fatty acid biosynthesis-, fatty acid oxidation-, and inflammation-associated factors. Besides, western blot analysis examined the expression of AMP-activated protein kinase (AMPK)/p38 signaling-associated factors. In palmitic acid (PA)-treated mice hepatocytes, RT-qPCR and western blot analysis examined Asprosin expression. Lipid accumulation, gluconeogenesis, fatty acid biosynthesis, fatty acid oxidation, and inflammation were appraised again.
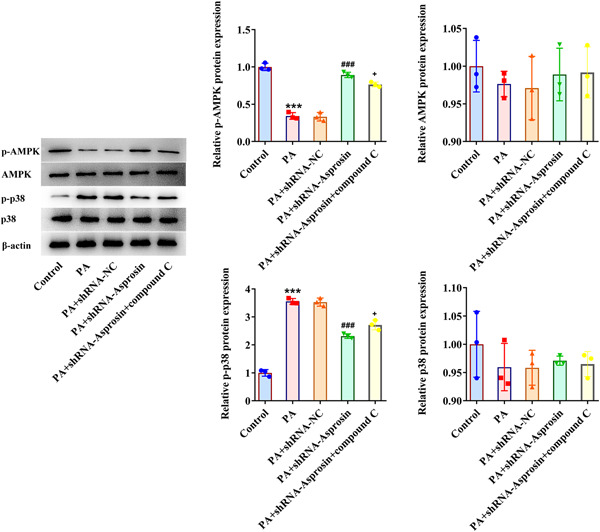
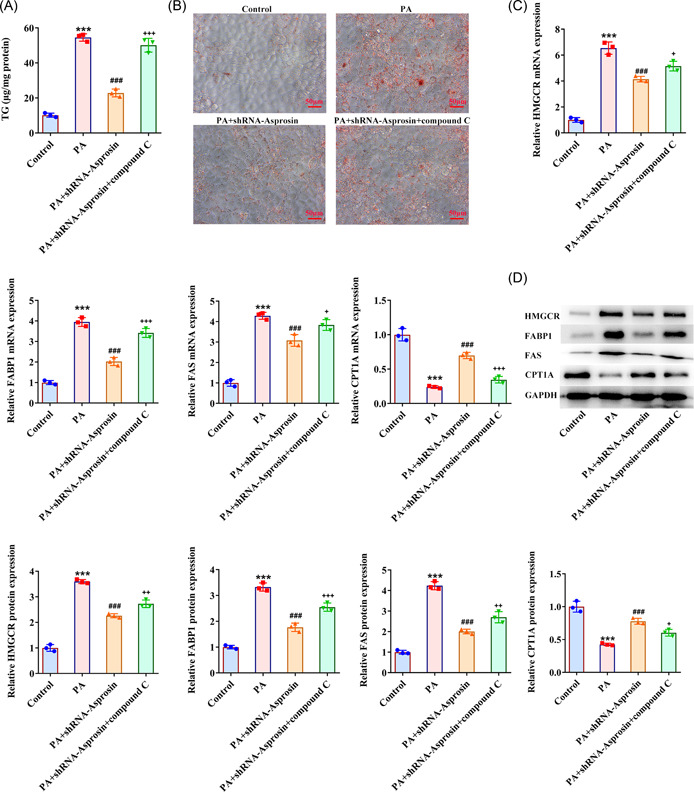
Results: Asprosin was overexpressed in the serum and liver tissues of NAFLD mice and PA-treated mice hepatocytes. Asprosin interference reduced mice body and liver weight, improved glucose tolerance and diminished liver injury in vivo. Asprosin knockdown alleviated lipid accumulation and inflammatory infiltration both in vitro and in vivo. Additionally, Asprosin absence activated AMPK/p38 signaling and AMPK inhibitor Compound C reversed the impacts of Asprosin on lipid accumulation and inflammatory response.
Conclusion: Collectively, Asprosin inhibition suppressed lipid accumulation and inflammation to obstruct NAFLD through AMPK/p38 signaling.
Keywords: AMPK signaling; Asprosin; inflammatory response; lipid accumulation; nonalcoholic fatty liver disease.
© 2023 The Authors. Immunity, Inflammation and Disease published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare that there is no conflicts of interest.
Figures









Similar articles
-
LB100 ameliorates nonalcoholic fatty liver disease via the AMPK/Sirt1 pathway.World J Gastroenterol. 2019 Dec 7;25(45):6607-6618. doi: 10.3748/wjg.v25.i45.6607. World J Gastroenterol. 2019. PMID: 31832001 Free PMC article.
-
Salvia-Nelumbinis naturalis improves lipid metabolism of NAFLD by regulating the SIRT1/AMPK signaling pathway.BMC Complement Med Ther. 2022 Aug 9;22(1):213. doi: 10.1186/s12906-022-03697-9. BMC Complement Med Ther. 2022. PMID: 35945571 Free PMC article.
-
Xiaozhi formula attenuates non-alcoholic fatty liver disease by regulating lipid metabolism via activation of AMPK and PPAR pathways.J Ethnopharmacol. 2024 Jul 15;329:118165. doi: 10.1016/j.jep.2024.118165. Epub 2024 Apr 7. J Ethnopharmacol. 2024. PMID: 38588984
-
Asperuloside alleviates lipid accumulation and inflammation in HFD-induced NAFLD via AMPK signaling pathway and NLRP3 inflammasome.Eur J Pharmacol. 2023 Mar 5;942:175504. doi: 10.1016/j.ejphar.2023.175504. Epub 2023 Jan 11. Eur J Pharmacol. 2023. PMID: 36641101
-
Targeting AMPK related signaling pathways: A feasible approach for natural herbal medicines to intervene non-alcoholic fatty liver disease.J Pharm Anal. 2025 Jan;15(1):101052. doi: 10.1016/j.jpha.2024.101052. Epub 2024 Aug 5. J Pharm Anal. 2025. PMID: 40034684 Free PMC article. Review.
Cited by
-
Effect of vitamin D and location of asprosin, spexin and meteorin-like antibodies in the liver of rats with isoproterenol-induced myocardial infarction.Naunyn Schmiedebergs Arch Pharmacol. 2025 Jun 28. doi: 10.1007/s00210-025-04409-z. Online ahead of print. Naunyn Schmiedebergs Arch Pharmacol. 2025. PMID: 40580311
-
Association between neutrophil-to-high-density lipoprotein cholesterol ratio and metabolic dysfunction-associated steatotic liver disease and liver fibrosis in the US population: a nationally representative cross-sectional study using NHANES data from 2017 to 2020.BMC Gastroenterol. 2024 Sep 5;24(1):300. doi: 10.1186/s12876-024-03394-6. BMC Gastroenterol. 2024. PMID: 39237899 Free PMC article.
-
Hepatic Olfr734 Deficiency Worsens Hepatic Glucose Metabolism and Induces MASLD in Mice.Nutrients. 2025 Jul 25;17(15):2426. doi: 10.3390/nu17152426. Nutrients. 2025. PMID: 40806011 Free PMC article.
-
MCC950 Ameliorates Diabetic Muscle Atrophy in Mice by Inhibition of Pyroptosis and Its Synergistic Effect with Aerobic Exercise.Molecules. 2024 Feb 4;29(3):712. doi: 10.3390/molecules29030712. Molecules. 2024. PMID: 38338456 Free PMC article.
-
Asprosin: its function as a novel endocrine factor in metabolic-related diseases.J Endocrinol Invest. 2024 Aug;47(8):1839-1850. doi: 10.1007/s40618-024-02360-z. Epub 2024 Apr 3. J Endocrinol Invest. 2024. PMID: 38568373 Review.
References
-
- Stefan N, Cusi K. A global view of the interplay between non‐alcoholic fatty liver disease and diabetes. Lancet Diab Endocrinol. 2022;10(4):284‐296. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
