

The effects of pathogenic and likely pathogenic variants for inherited hemostasis disorders in 140 214 UK Biobank participants
- PMID: 37647632
- PMCID: PMC10733830
- DOI: 10.1182/blood.2023020118
The effects of pathogenic and likely pathogenic variants for inherited hemostasis disorders in 140 214 UK Biobank participants
Abstract
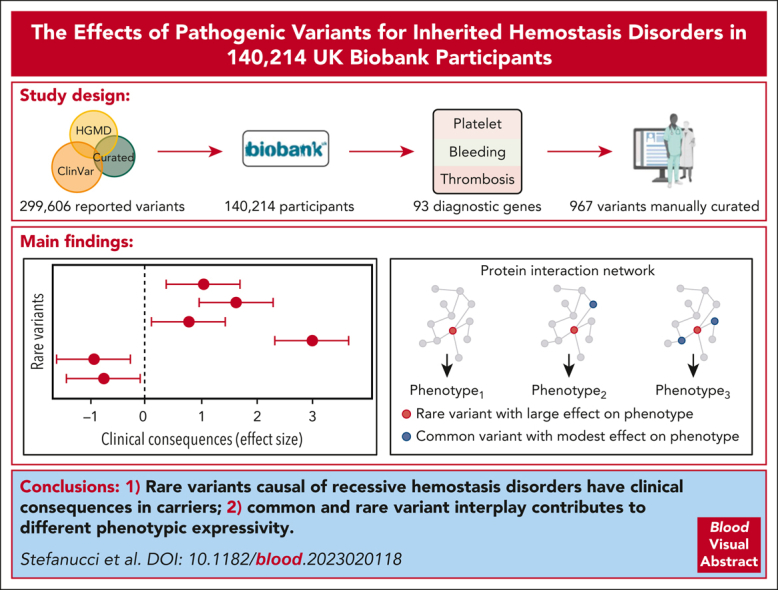
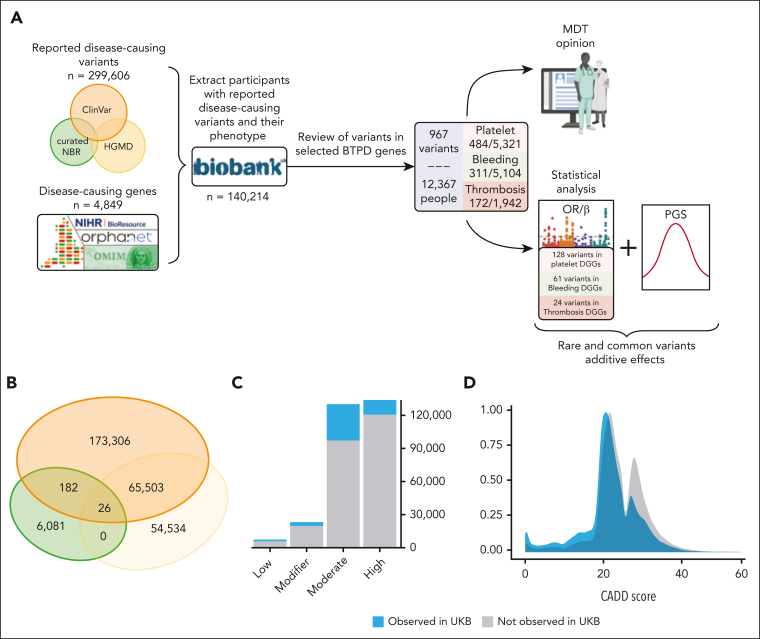
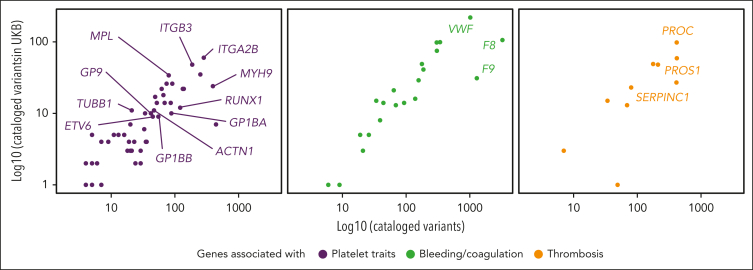
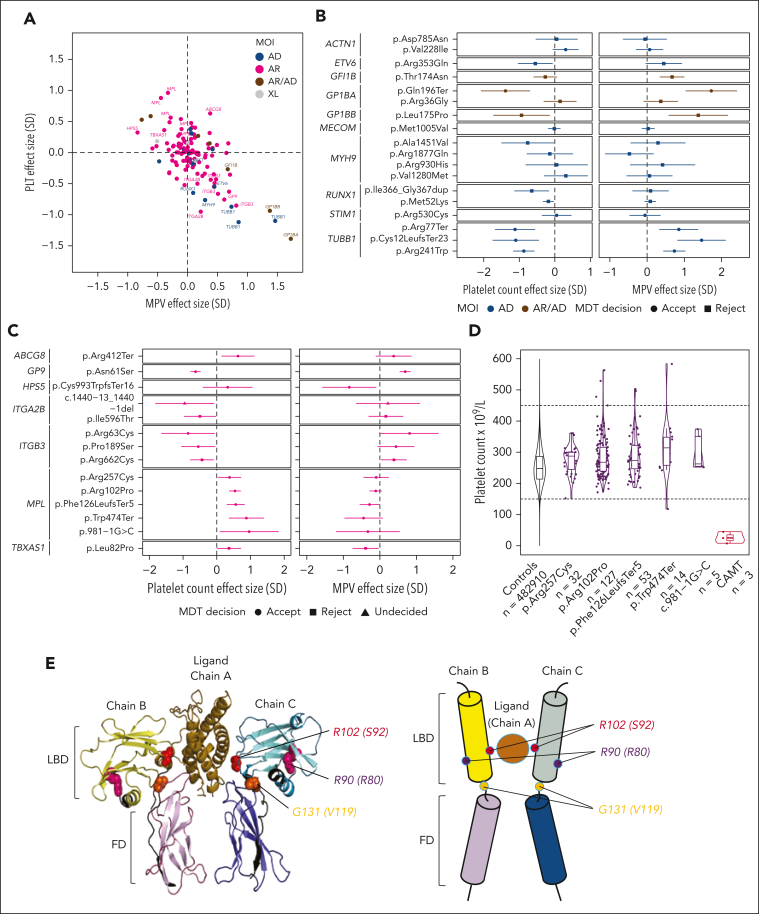
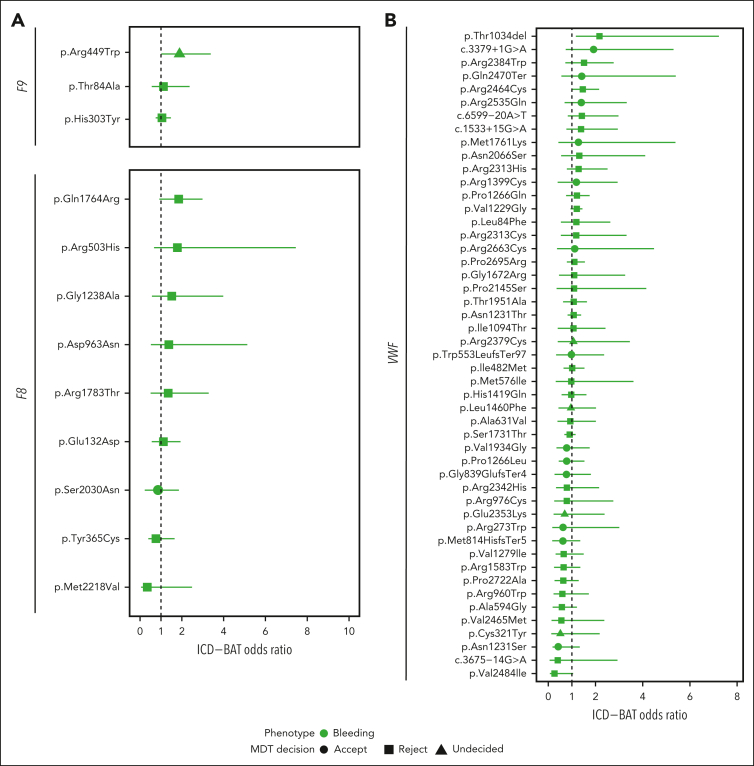
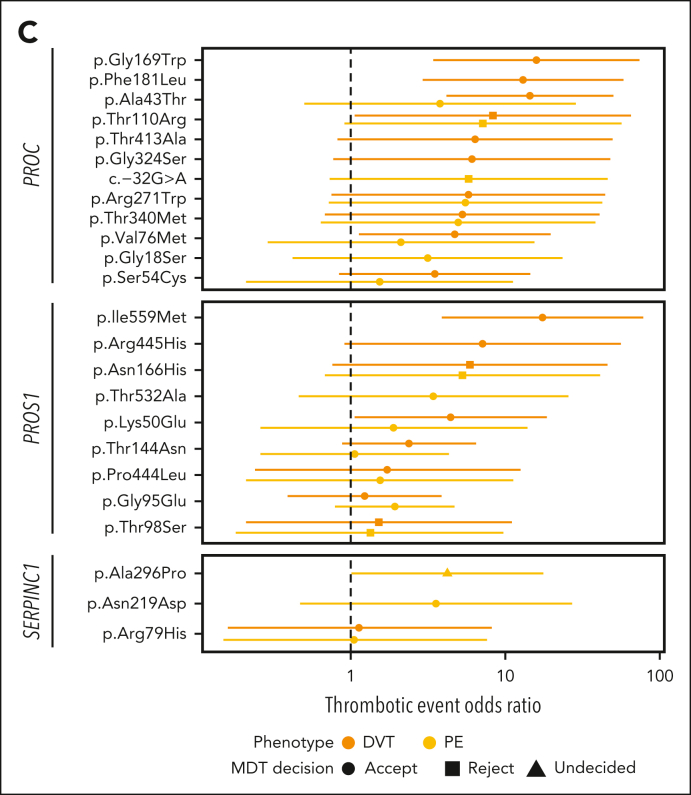
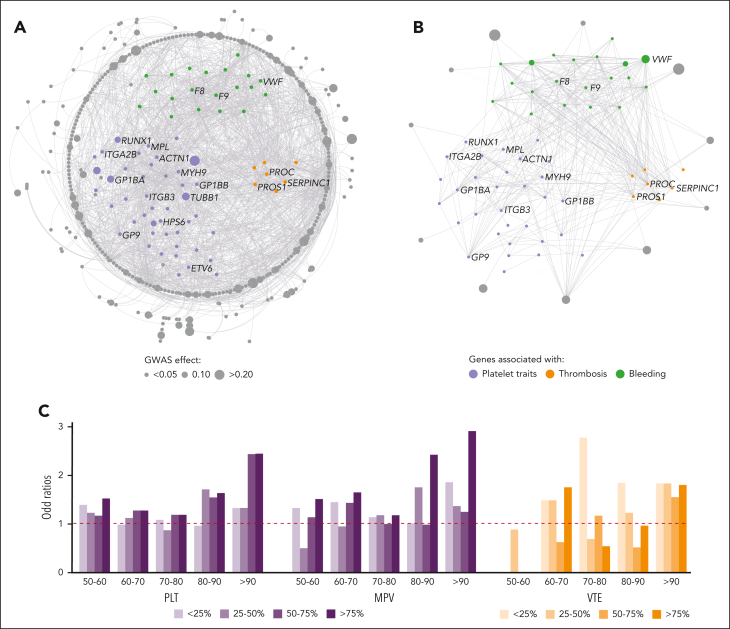
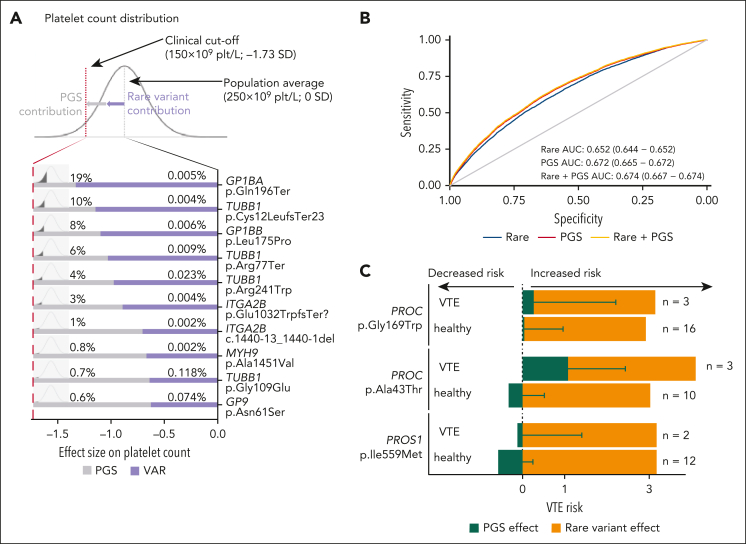
Rare genetic diseases affect millions, and identifying causal DNA variants is essential for patient care. Therefore, it is imperative to estimate the effect of each independent variant and improve their pathogenicity classification. Our study of 140 214 unrelated UK Biobank (UKB) participants found that each of them carries a median of 7 variants previously reported as pathogenic or likely pathogenic. We focused on 967 diagnostic-grade gene (DGG) variants for rare bleeding, thrombotic, and platelet disorders (BTPDs) observed in 12 367 UKB participants. By association analysis, for a subset of these variants, we estimated effect sizes for platelet count and volume, and odds ratios for bleeding and thrombosis. Variants causal of some autosomal recessive platelet disorders revealed phenotypic consequences in carriers. Loss-of-function variants in MPL, which cause chronic amegakaryocytic thrombocytopenia if biallelic, were unexpectedly associated with increased platelet counts in carriers. We also demonstrated that common variants identified by genome-wide association studies (GWAS) for platelet count or thrombosis risk may influence the penetrance of rare variants in BTPD DGGs on their associated hemostasis disorders. Network-propagation analysis applied to an interactome of 18 410 nodes and 571 917 edges showed that GWAS variants with large effect sizes are enriched in DGGs and their first-order interactors. Finally, we illustrate the modifying effect of polygenic scores for platelet count and thrombosis risk on disease severity in participants carrying rare variants in TUBB1 or PROC and PROS1, respectively. Our findings demonstrate the power of association analyses using large population datasets in improving pathogenicity classifications of rare variants.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: O.S.B., Q.W., K.C., P.P., K.M., and S.P. are current AstraZeneca employees and/or stockholders. L. Sun is a full-time employee at Regeneron Genetics Center, LLC. The remaining authors declare no competing financial interests.
The current affiliation for P.P. is R&D Data Office, Data Science and Artificial Intelligence, BioPharmaceuticals R&D, AstraZeneca, Cambridge, United Kingdom.
The current affiliation for K.M. is Centre for Genomics Research, Discovery Sciences, BioPharmaceuticals R&D, AstraZeneca, Cambridge, United Kingdom.
A complete list of the members of the NIHR BioResource Consortium appears in the supplemental Material.
Figures
Comment in
-
From black and white to fifty shades of grey.Blood. 2023 Dec 14;142(24):2037-2038. doi: 10.1182/blood.2023022171. Blood. 2023. PMID: 38095926 No abstract available.
References
-
- Ferreira CR. The burden of rare diseases. Am J Med Genet A. 2019;179(6):885–892. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
