

Gut microbial carbohydrate metabolism contributes to insulin resistance
- PMID: 37648852
- PMCID: PMC10499599
- DOI: 10.1038/s41586-023-06466-x
Gut microbial carbohydrate metabolism contributes to insulin resistance
Abstract
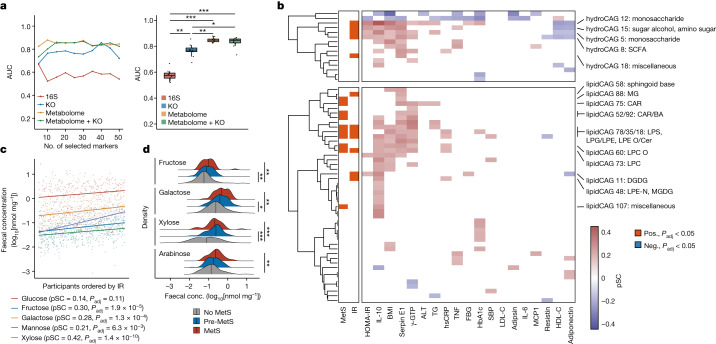
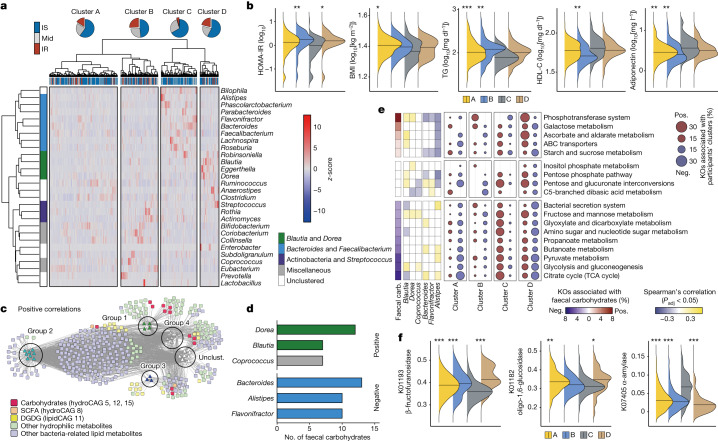
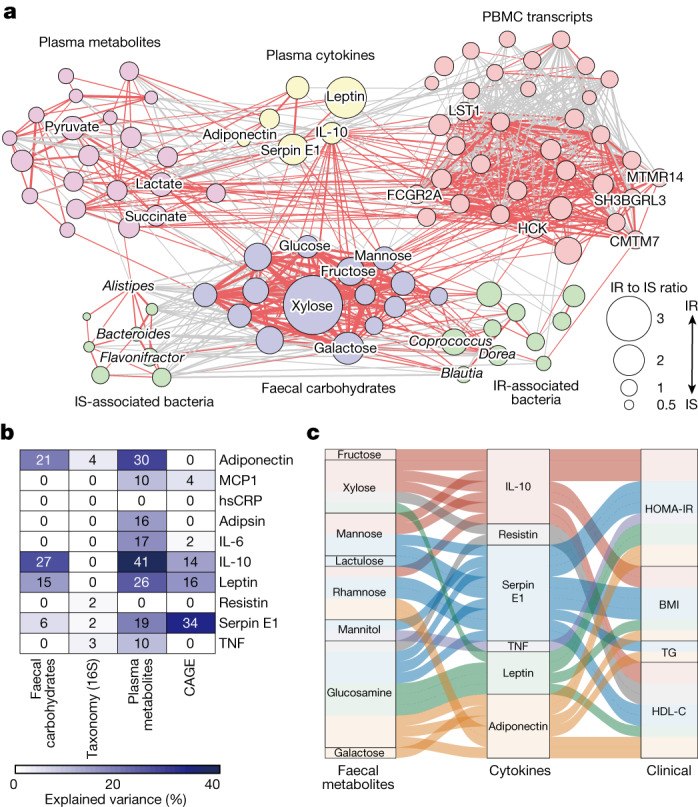
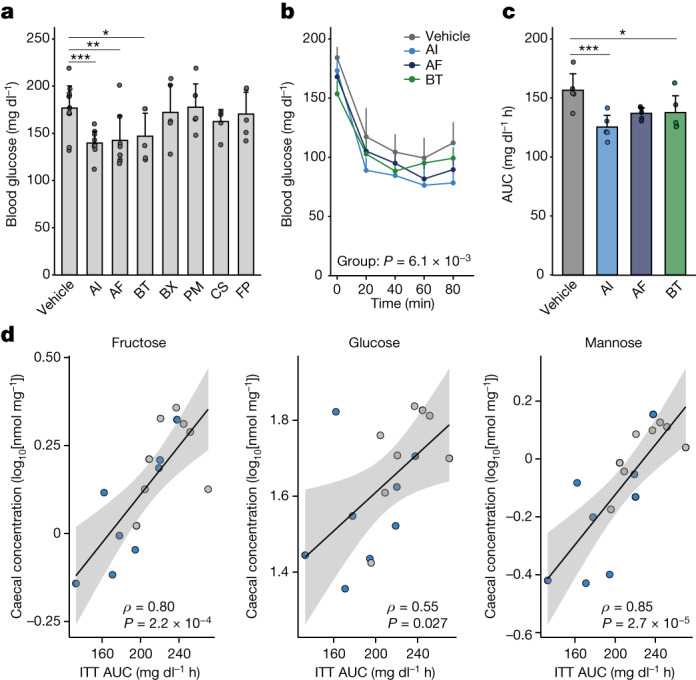
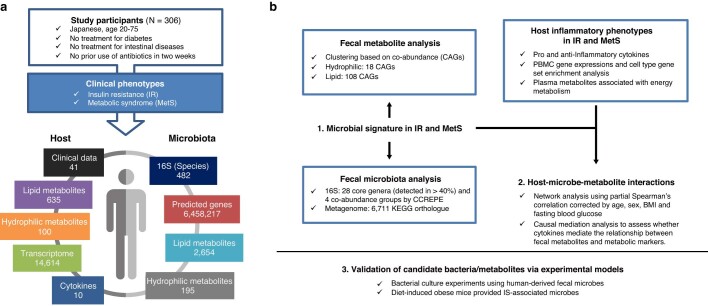
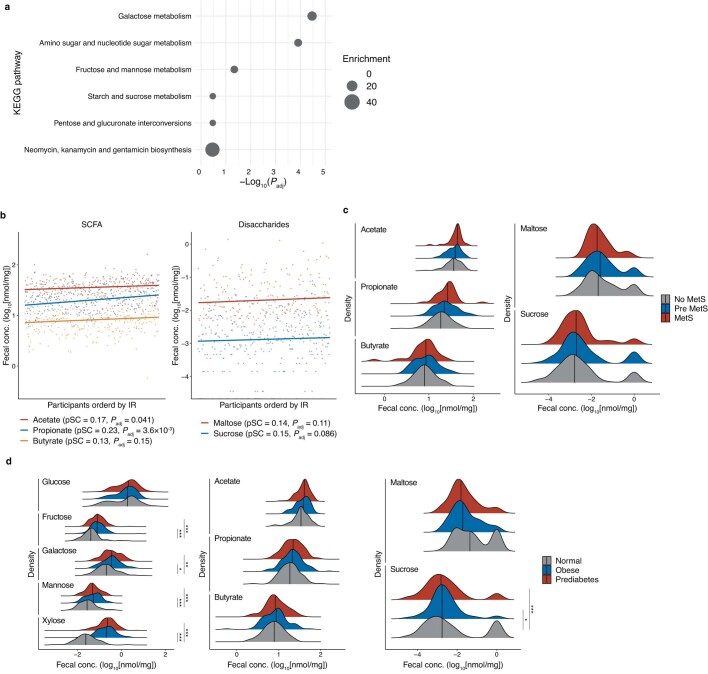
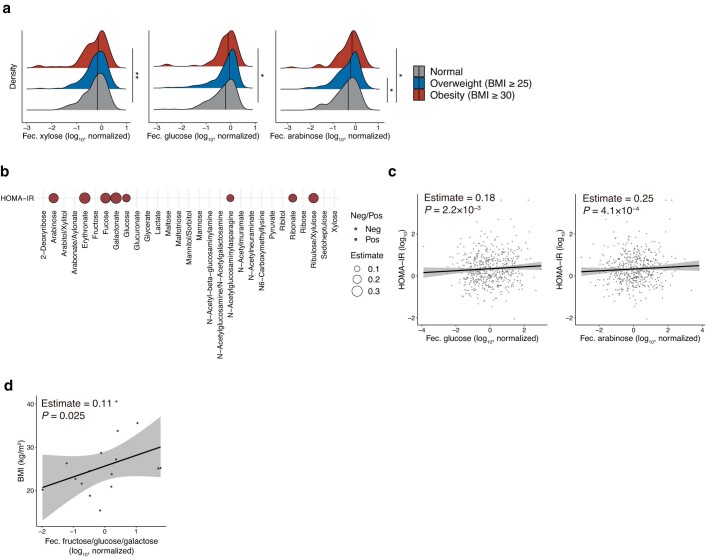
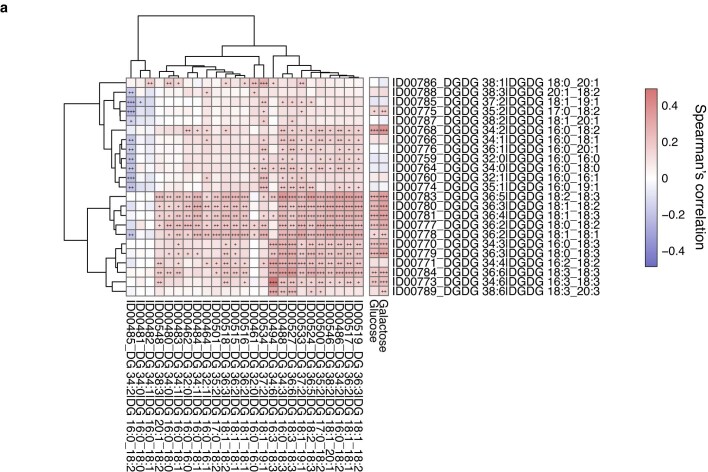
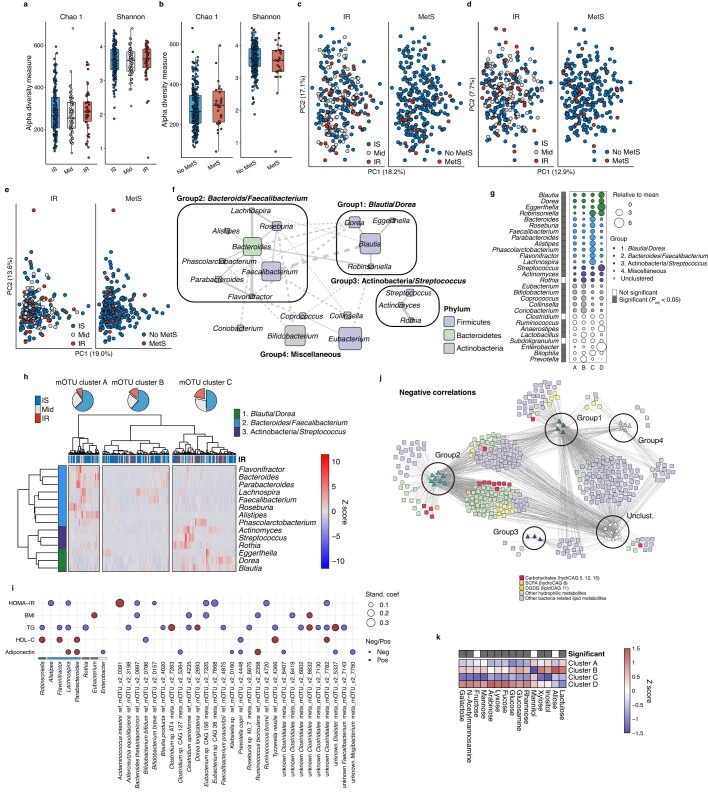
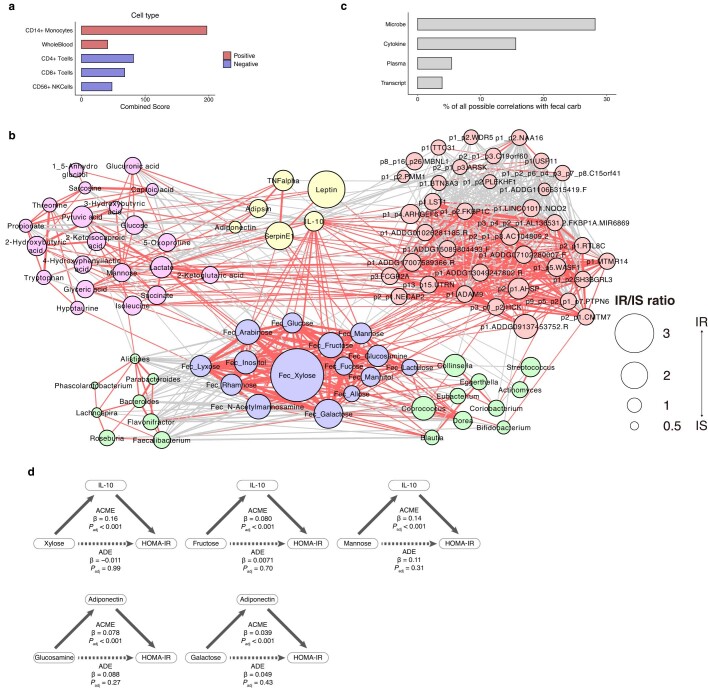
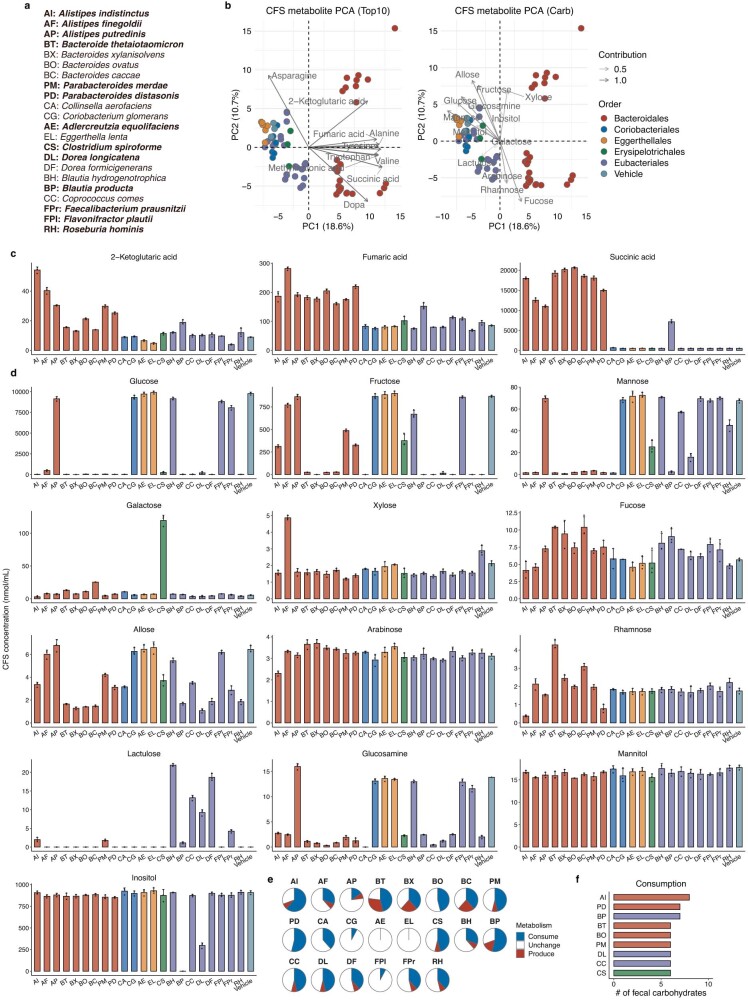
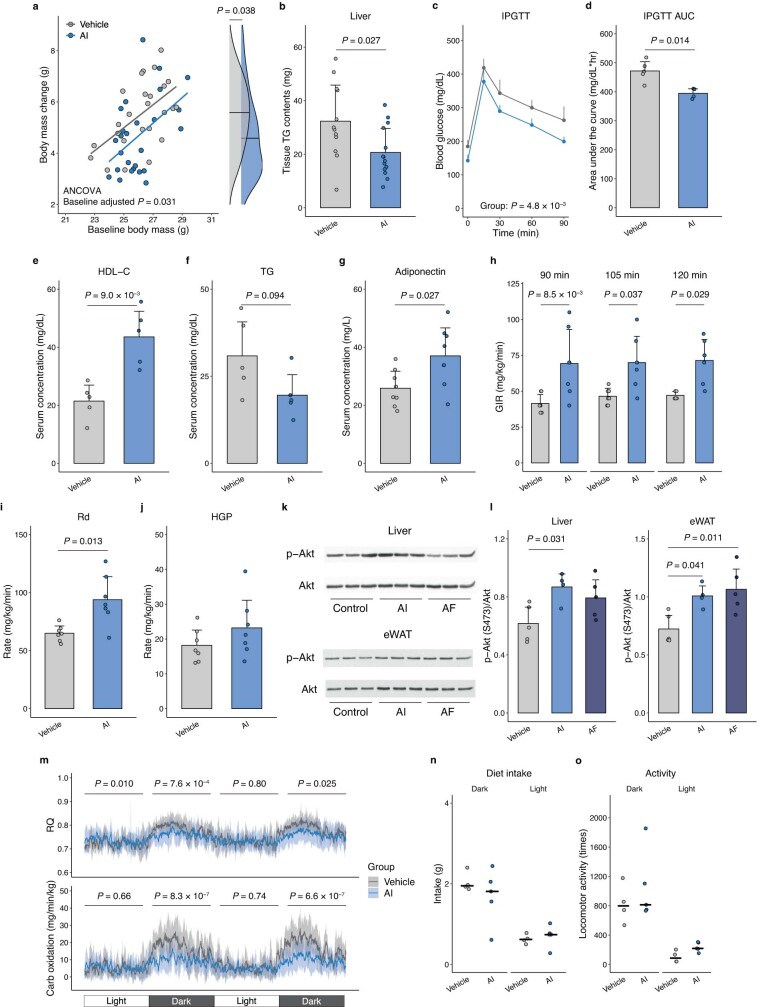
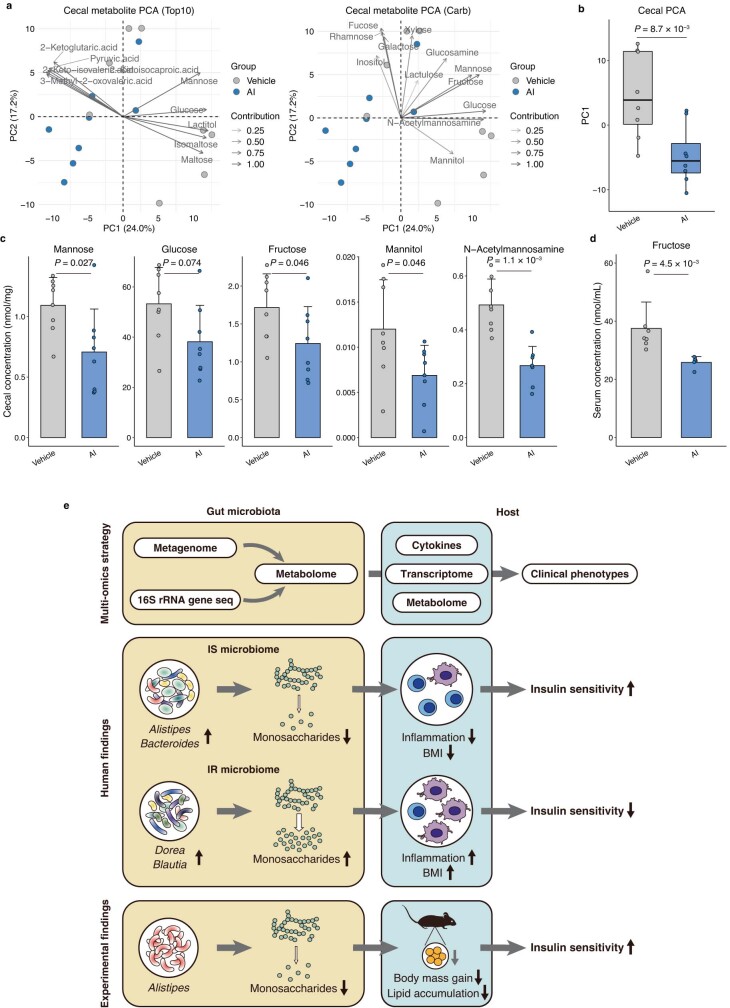
Insulin resistance is the primary pathophysiology underlying metabolic syndrome and type 2 diabetes1,2. Previous metagenomic studies have described the characteristics of gut microbiota and their roles in metabolizing major nutrients in insulin resistance3-9. In particular, carbohydrate metabolism of commensals has been proposed to contribute up to 10% of the host's overall energy extraction10, thereby playing a role in the pathogenesis of obesity and prediabetes3,4,6. Nevertheless, the underlying mechanism remains unclear. Here we investigate this relationship using a comprehensive multi-omics strategy in humans. We combine unbiased faecal metabolomics with metagenomics, host metabolomics and transcriptomics data to profile the involvement of the microbiome in insulin resistance. These data reveal that faecal carbohydrates, particularly host-accessible monosaccharides, are increased in individuals with insulin resistance and are associated with microbial carbohydrate metabolisms and host inflammatory cytokines. We identify gut bacteria associated with insulin resistance and insulin sensitivity that show a distinct pattern of carbohydrate metabolism, and demonstrate that insulin-sensitivity-associated bacteria ameliorate host phenotypes of insulin resistance in a mouse model. Our study, which provides a comprehensive view of the host-microorganism relationships in insulin resistance, reveals the impact of carbohydrate metabolism by microbiota, suggesting a potential therapeutic target for ameliorating insulin resistance.
© 2023. The Author(s).
Conflict of interest statement
T.T., Y.N., W.S. and H.O. are listed as the inventors on a patent regarding the metabolic effects of gut bacteria identified by a human cohort. The other authors declare no competing interests.
Figures

Comment in
-
Microbial metabolism linked to insulin resistance.Nat Rev Endocrinol. 2023 Nov;19(11):620. doi: 10.1038/s41574-023-00903-7. Nat Rev Endocrinol. 2023. PMID: 37696918 No abstract available.
-
Microbial stars: shedding light on gut microbes' role in insulin resistance and innovative diabetes therapies.Gut Microbes. 2024 Jan-Dec;16(1):2307581. doi: 10.1080/19490976.2024.2307581. Epub 2024 Jan 26. Gut Microbes. 2024. PMID: 38277136 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
