

LTP induction by structural rather than enzymatic functions of CaMKII
- PMID: 37648853
- PMCID: PMC10482691
- DOI: 10.1038/s41586-023-06465-y
LTP induction by structural rather than enzymatic functions of CaMKII
Abstract
Learning and memory are thought to require hippocampal long-term potentiation (LTP), and one of the few central dogmas of molecular neuroscience that has stood undisputed for more than three decades is that LTP induction requires enzymatic activity of the Ca2+/calmodulin-dependent protein kinase II (CaMKII)1-3. However, as we delineate here, the experimental evidence is surprisingly far from conclusive. All previous interventions inhibiting enzymatic CaMKII activity and LTP4-8 also interfere with structural CaMKII roles, in particular binding to the NMDA-type glutamate receptor subunit GluN2B9-14. Thus, we here characterized and utilized complementary sets of new opto-/pharmaco-genetic tools to distinguish between enzymatic and structural CaMKII functions. Several independent lines of evidence demonstrated LTP induction by a structural function of CaMKII rather than by its enzymatic activity. The sole contribution of kinase activity was autoregulation of this structural role via T286 autophosphorylation, which explains why this distinction has been elusive for decades. Directly initiating the structural function in a manner that circumvented this T286 role was sufficient to elicit robust LTP, even when enzymatic CaMKII activity was blocked.
© 2023. The Author(s).
Conflict of interest statement
The Regents of the University of Colorado have filed a provisional patent application, with inventors J.E.T., M.E.L., N.L.R. and K.U.B., capturing the findings reported in this manuscript. K.U.B. is a co-founder and board member of Neurexis Therapeutics, a company that seeks to develop a CaMKII inhibitor into a therapeutic drug for cerebral ischaemia. The other authors declare no competing interests.
Figures
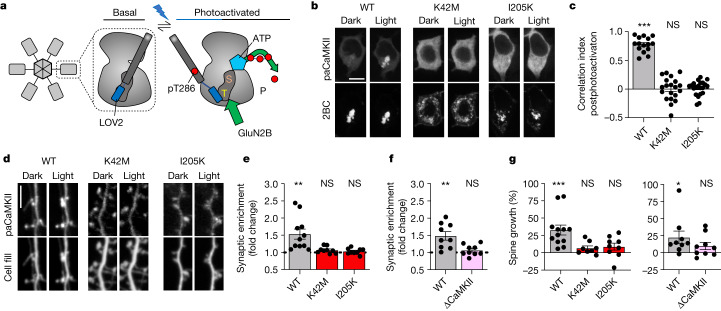
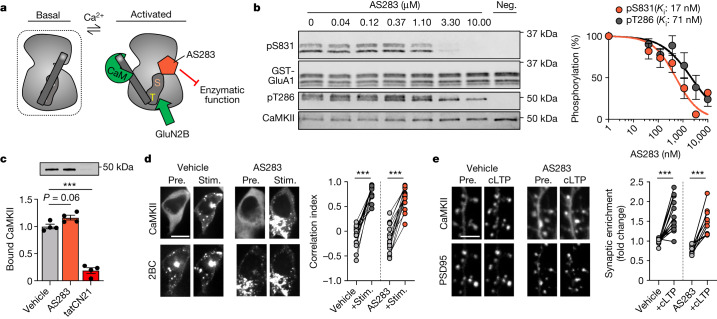


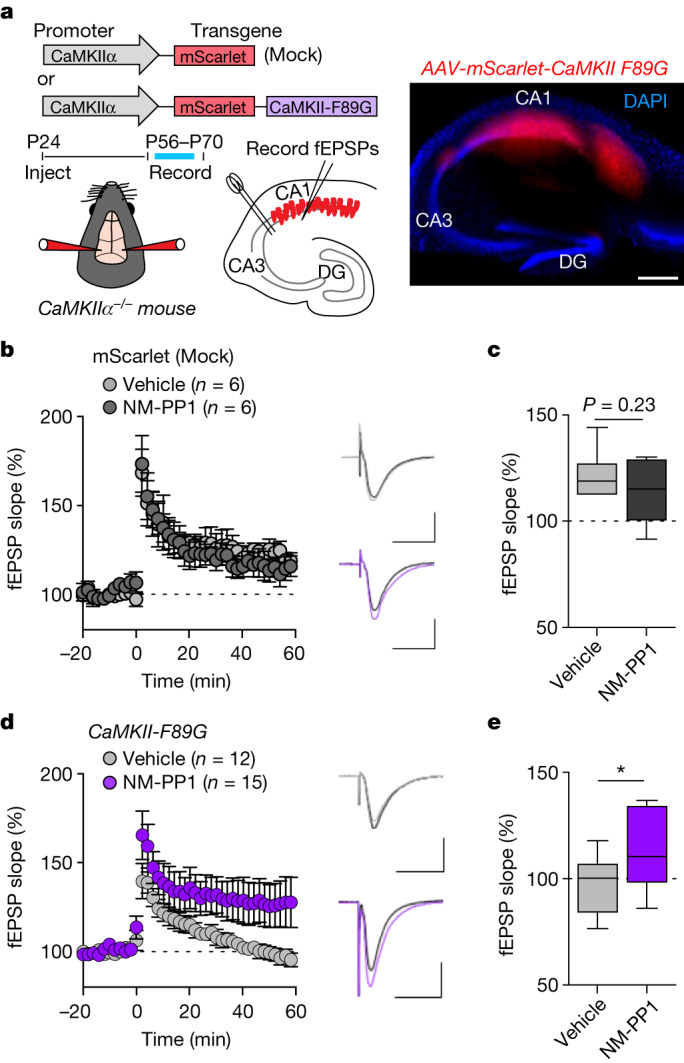
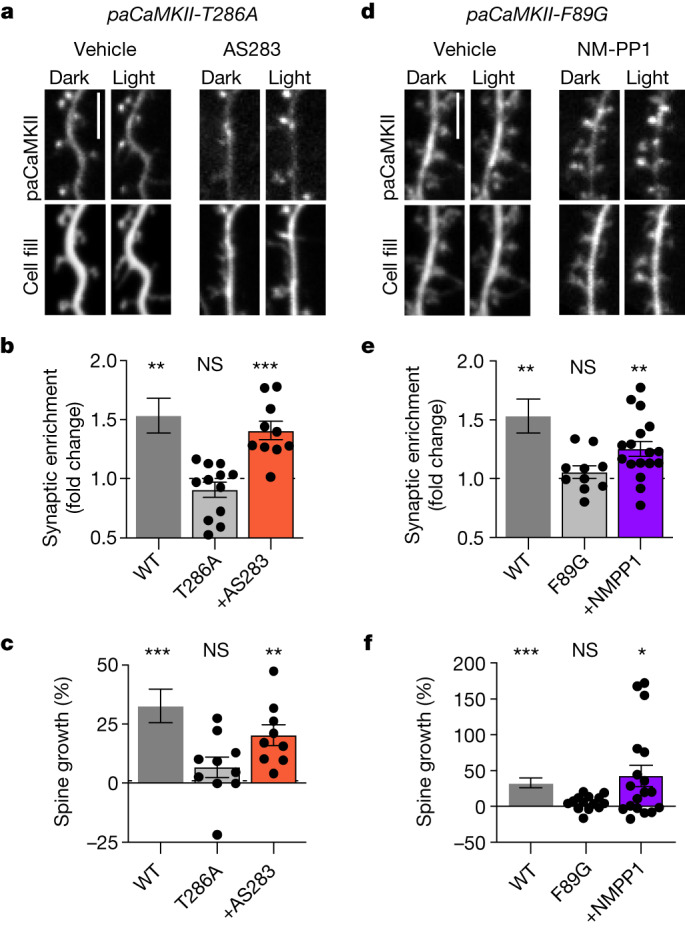






Comment in
-
A structural function for CaMKII.Nat Rev Neurosci. 2023 Nov;24(11):654. doi: 10.1038/s41583-023-00754-1. Nat Rev Neurosci. 2023. PMID: 37770621 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- F31 MH125510/MH/NIMH NIH HHS/United States
- F31 AG084197/AG/NIA NIH HHS/United States
- F31 NS129254/NS/NINDS NIH HHS/United States
- F31 AG062160/AG/NIA NIH HHS/United States
- R01 MH116901/MH/NIMH NIH HHS/United States
- T32 GM007635/GM/NIGMS NIH HHS/United States
- R01 NS040701/NS/NINDS NIH HHS/United States
- R01 NS118786/NS/NINDS NIH HHS/United States
- R01 AG067713/AG/NIA NIH HHS/United States
- R01 NS081248/NS/NINDS NIH HHS/United States
- F31 AG069458/AG/NIA NIH HHS/United States
- R01 NS080851/NS/NINDS NIH HHS/United States
- R01 NS110383/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
