

Epitope editing enables targeted immunotherapy of acute myeloid leukaemia
- PMID: 37648862
- PMCID: PMC10499609
- DOI: 10.1038/s41586-023-06496-5
Epitope editing enables targeted immunotherapy of acute myeloid leukaemia
Abstract
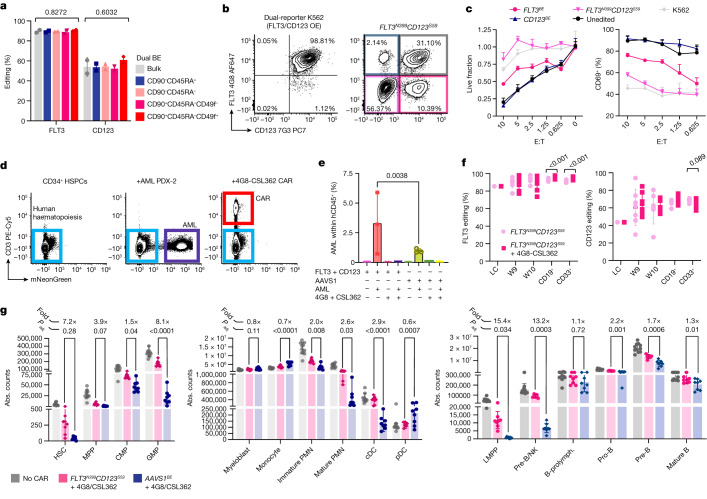
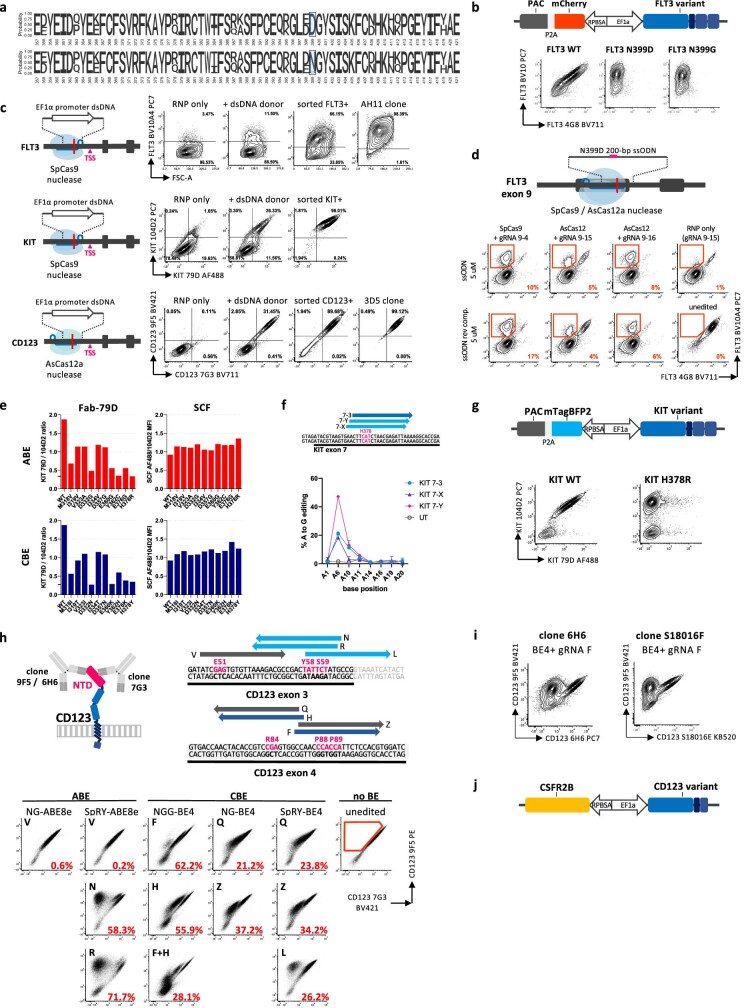
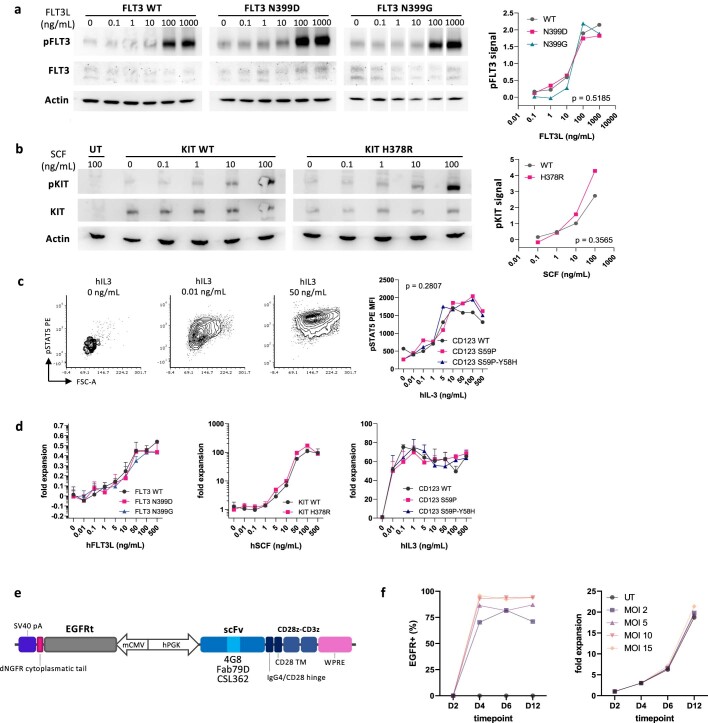
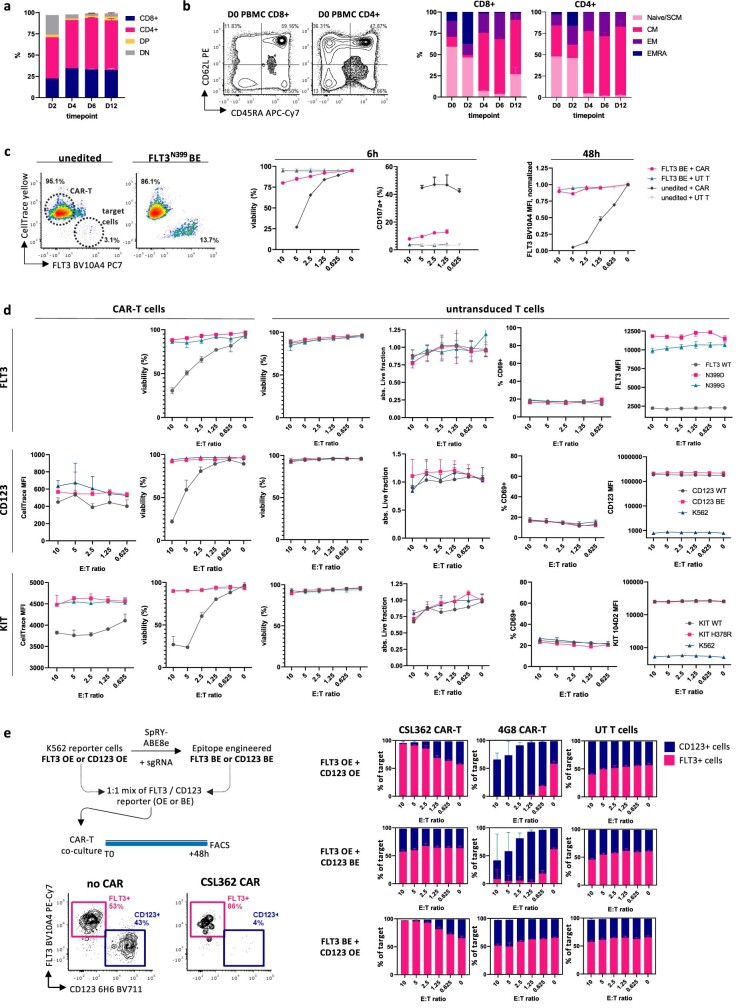
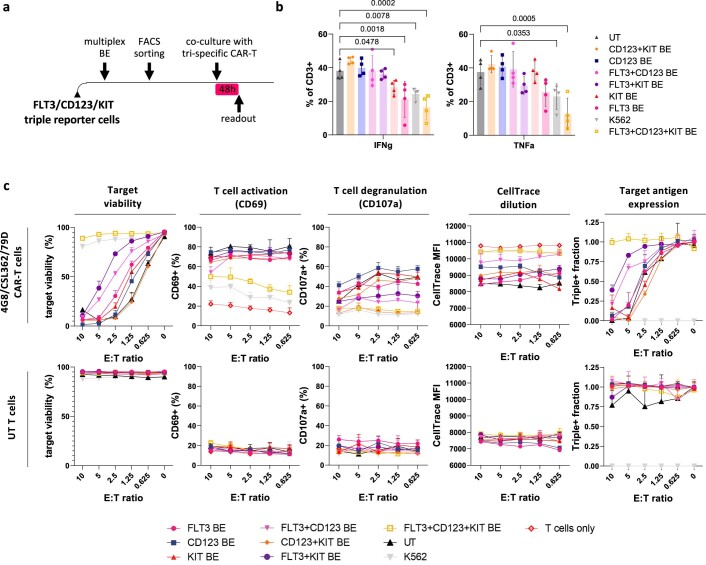
Despite the considerable efficacy observed when targeting a dispensable lineage antigen, such as CD19 in B cell acute lymphoblastic leukaemia1,2, the broader applicability of adoptive immunotherapies is hampered by the absence of tumour-restricted antigens3-5. Acute myeloid leukaemia immunotherapies target genes expressed by haematopoietic stem/progenitor cells (HSPCs) or differentiated myeloid cells, resulting in intolerable on-target/off-tumour toxicity. Here we show that epitope engineering of donor HSPCs used for bone marrow transplantation endows haematopoietic lineages with selective resistance to chimeric antigen receptor (CAR) T cells or monoclonal antibodies, without affecting protein function or regulation. This strategy enables the targeting of genes that are essential for leukaemia survival regardless of shared expression on HSPCs, reducing the risk of tumour immune escape. By performing epitope mapping and library screenings, we identified amino acid changes that abrogate the binding of therapeutic monoclonal antibodies targeting FLT3, CD123 and KIT, and optimized a base-editing approach to introduce them into CD34+ HSPCs, which retain long-term engraftment and multilineage differentiation ability. After CAR T cell treatment, we confirmed resistance of epitope-edited haematopoiesis and concomitant eradication of patient-derived acute myeloid leukaemia xenografts. Furthermore, we show that multiplex epitope engineering of HSPCs is feasible and enables more effective immunotherapies against multiple targets without incurring overlapping off-tumour toxicities. We envision that this approach will provide opportunities to treat relapsed/refractory acute myeloid leukaemia and enable safer non-genotoxic conditioning.
© 2023. The Author(s).
Conflict of interest statement
G.C. and P.G. are listed as inventors on patent applications related to this work that are owned and managed by the Boston Children’s Hospital and Dana-Farber Cancer Institute. S.A.A. has been a consultant and/or shareholder for Neomorph Inc, Imago Biosciences, Cyteir Therapeutics, C4 Therapeutics, Nimbus Therapeutics and Accent Therapeutics. S.A.A. has received research support from Janssen and Syndax. S.A.A. is named as an inventor on a patent application related to MENIN inhibition WO/2017/132398A1. J.A.M. is a founder, equity holder and advisor to Entact Bio, serves on the scientific advisory board of 908 Devices and receives sponsored research funding from Vertex, AstraZeneca, Taiho, Springworks and TUO Therapeutics. The other authors declare no competing interests.
Figures
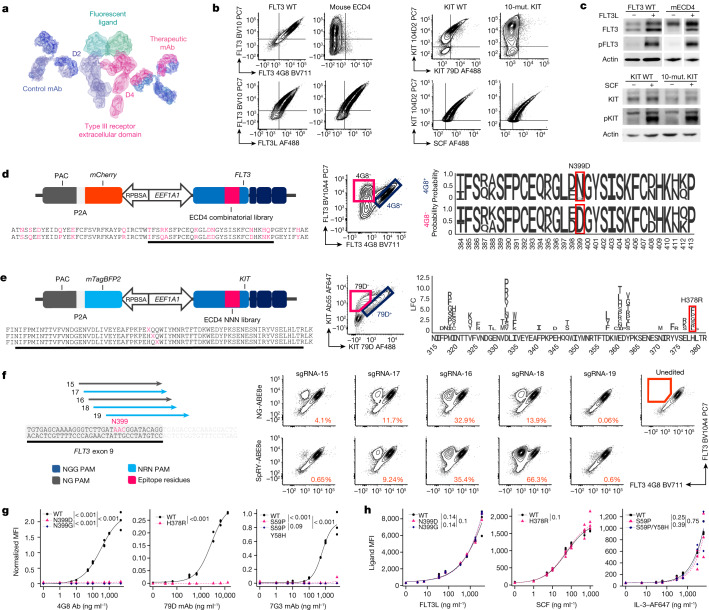
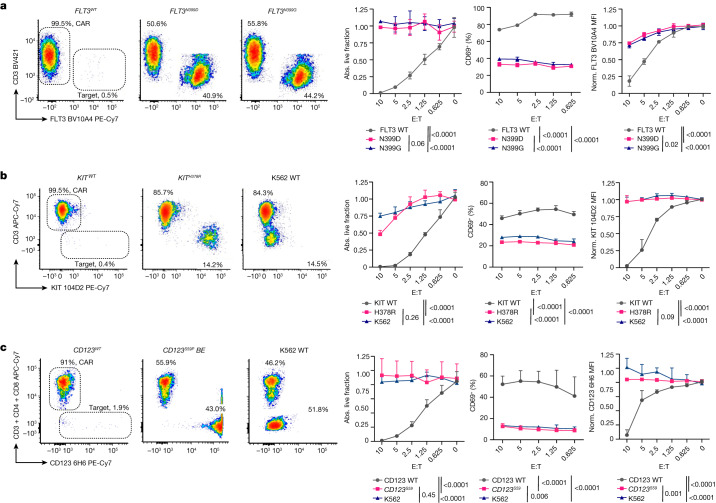
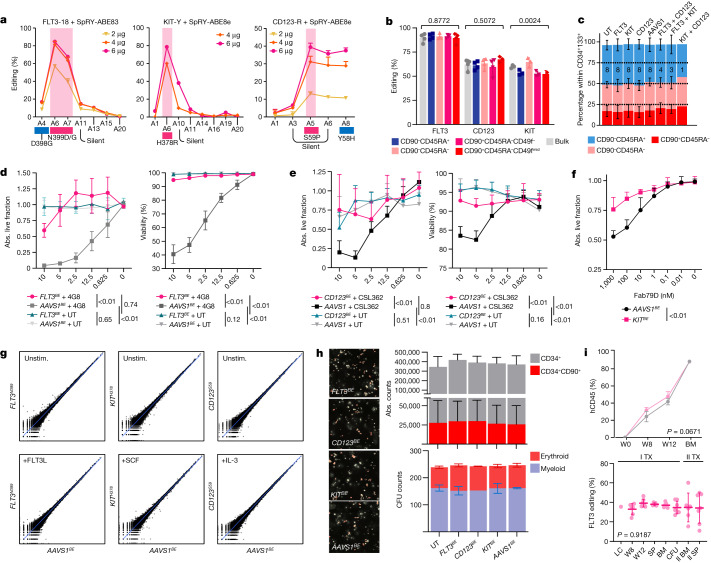
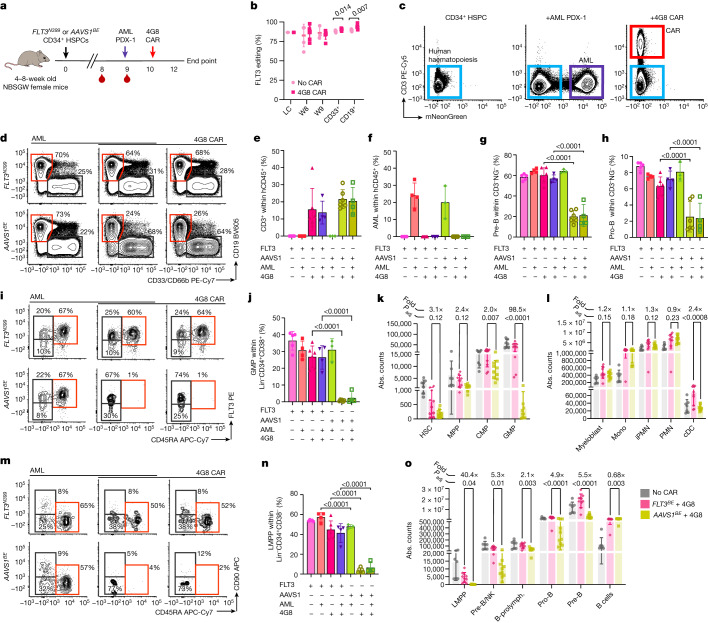
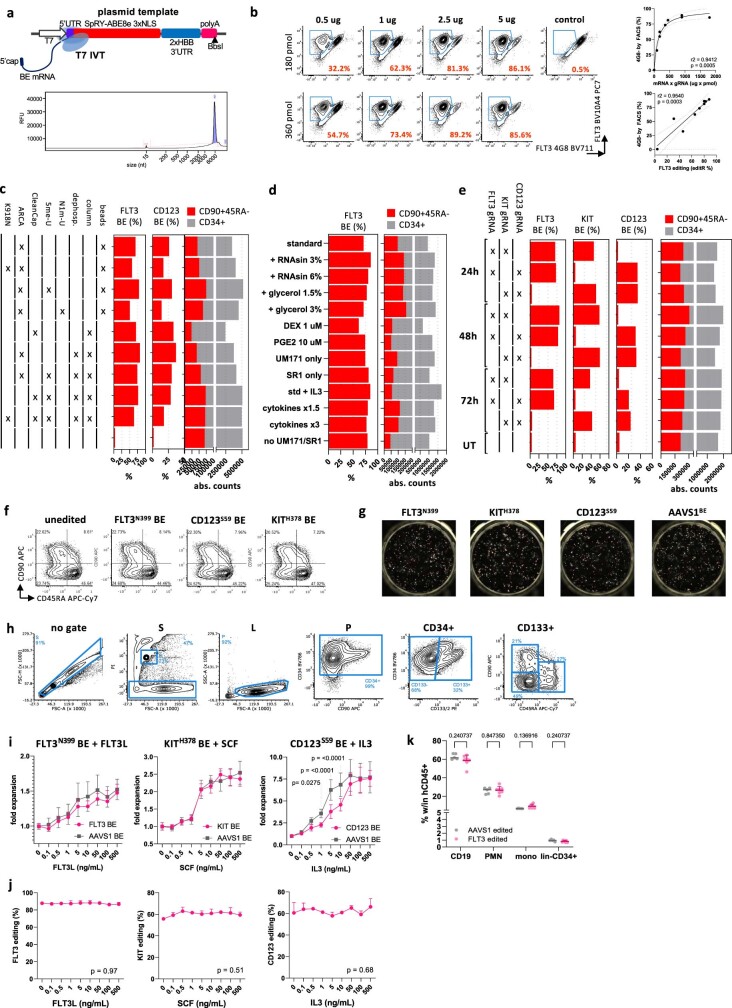
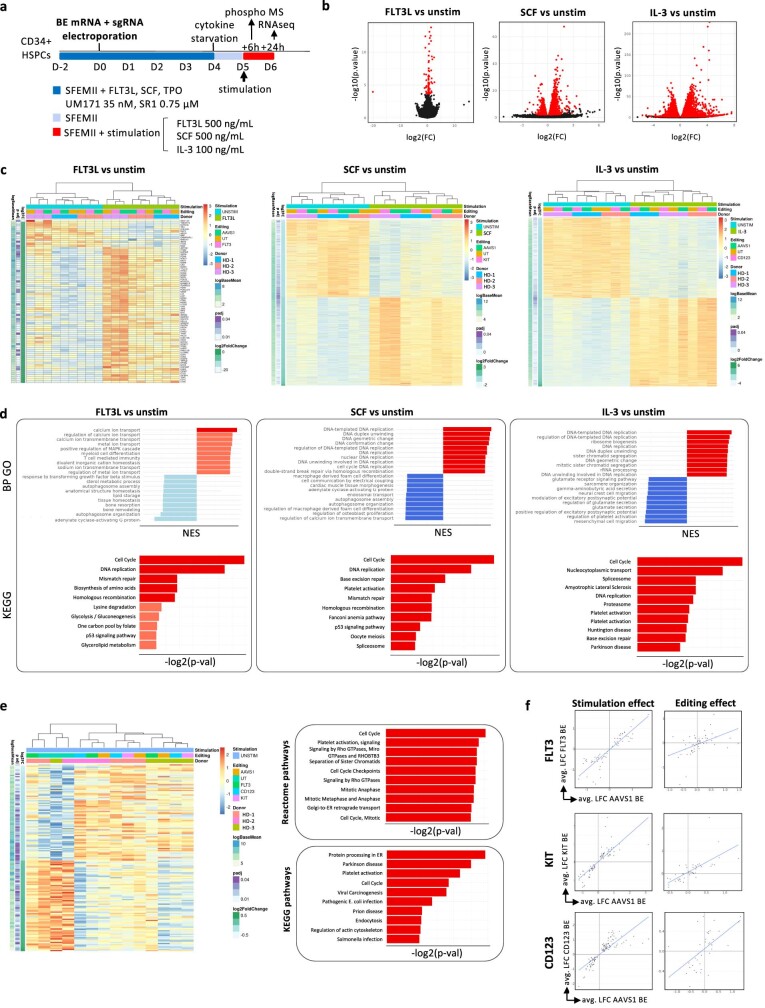
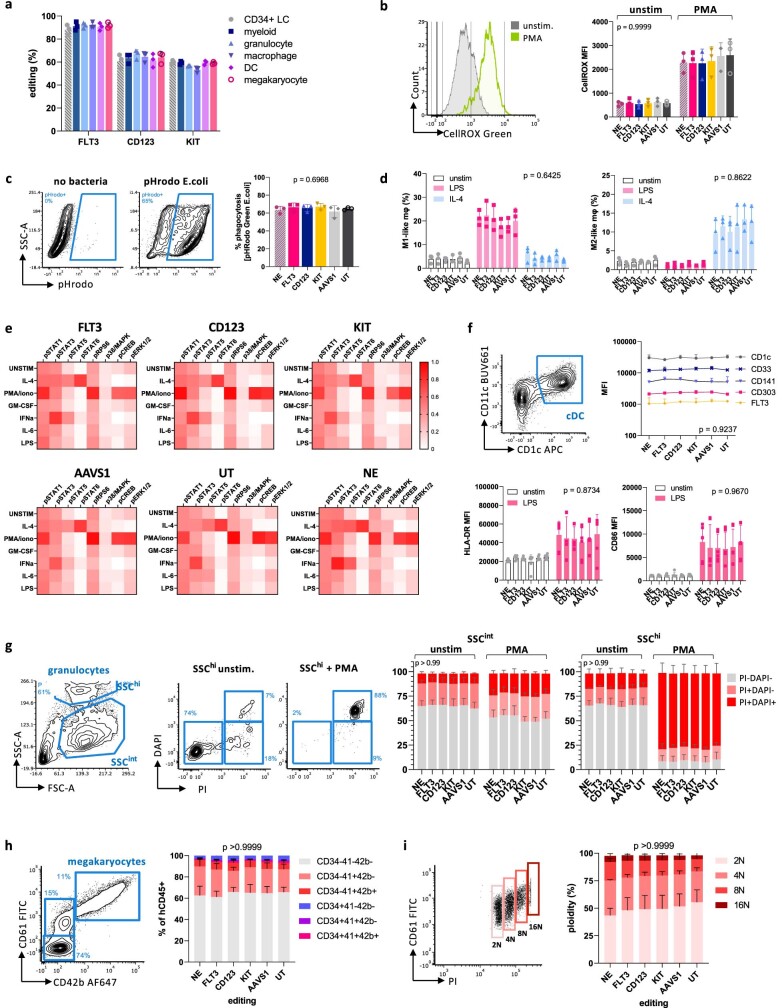
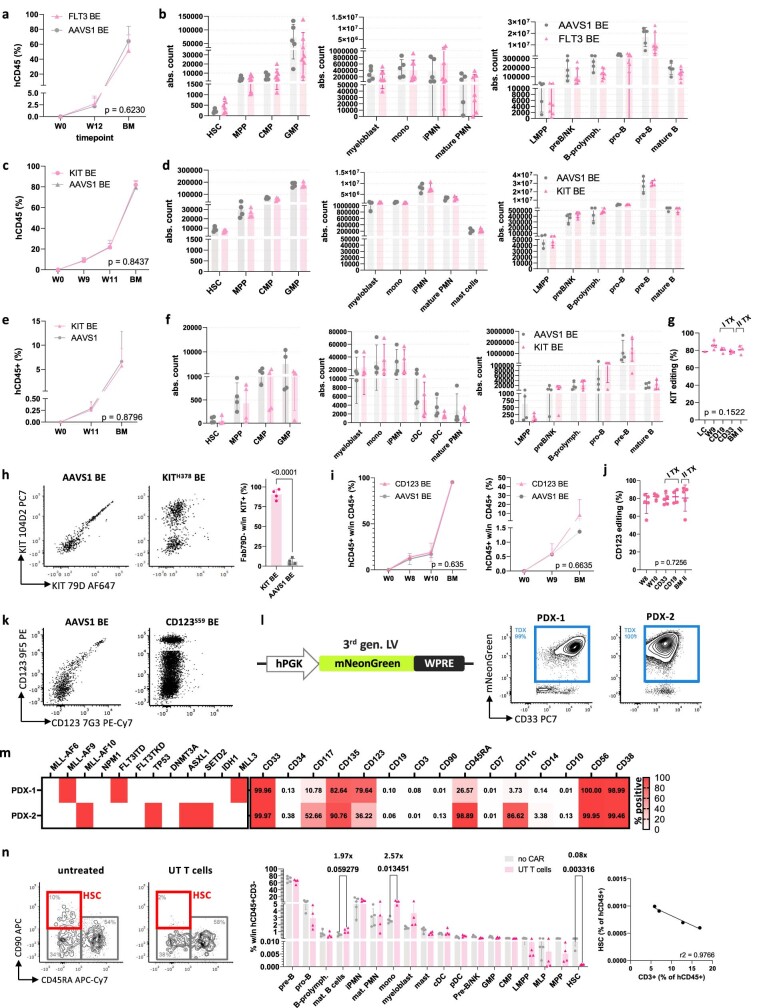
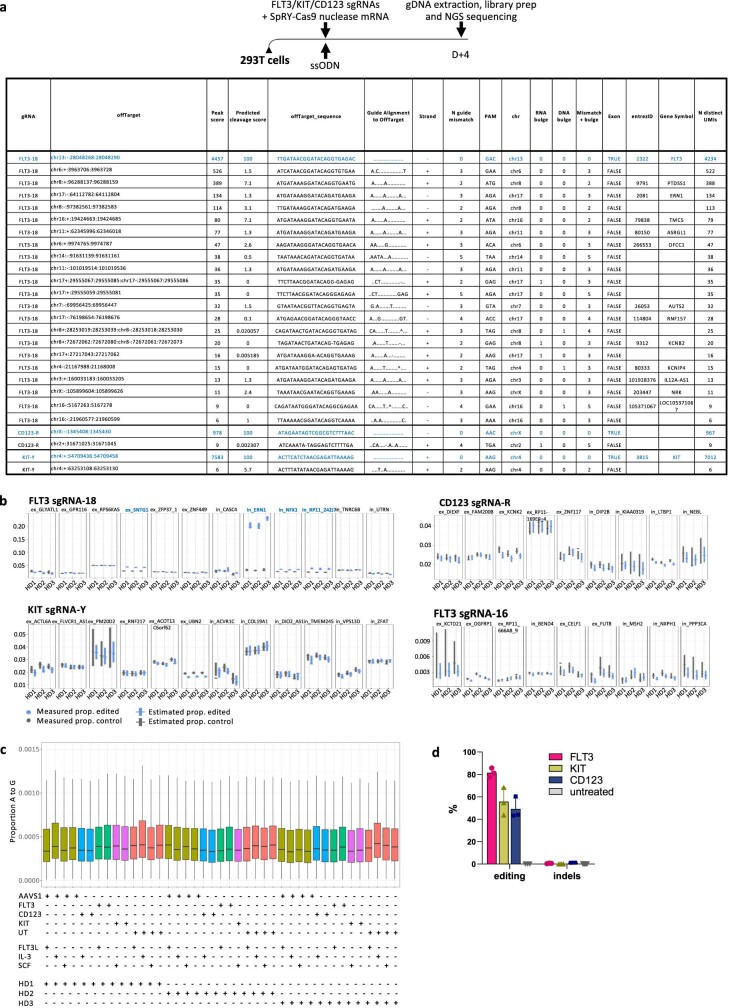
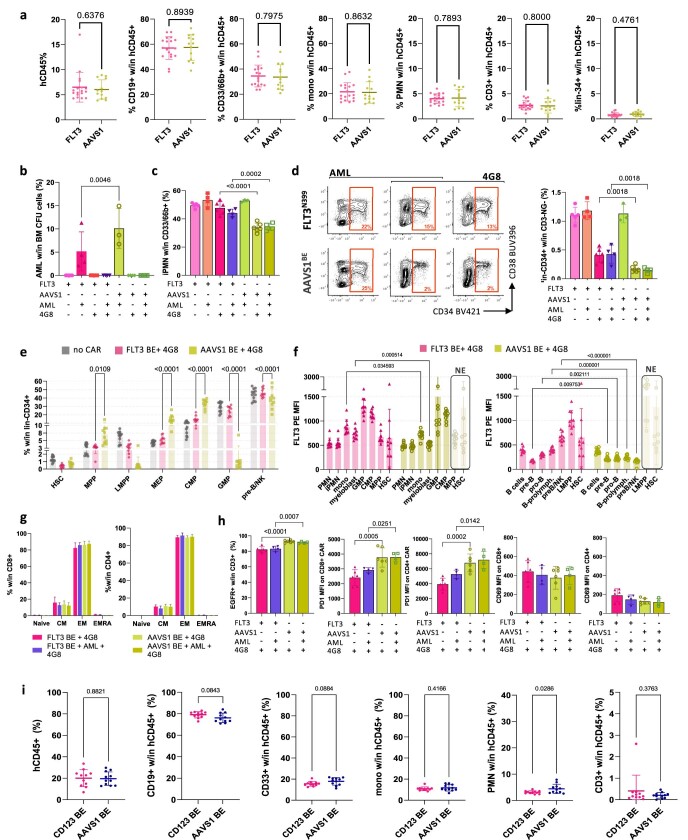
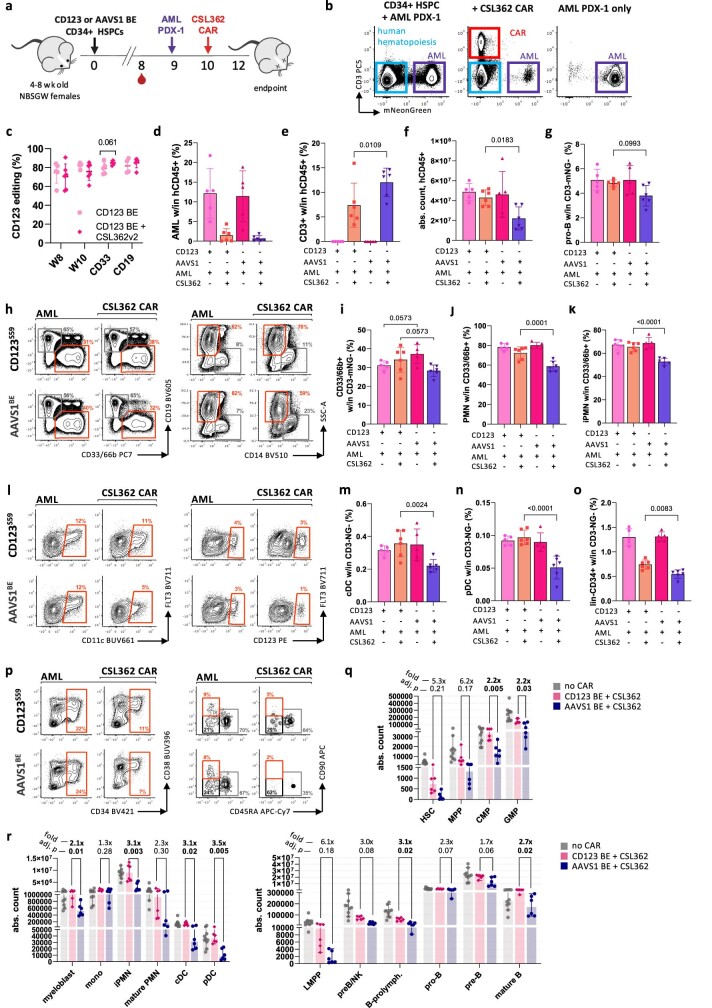
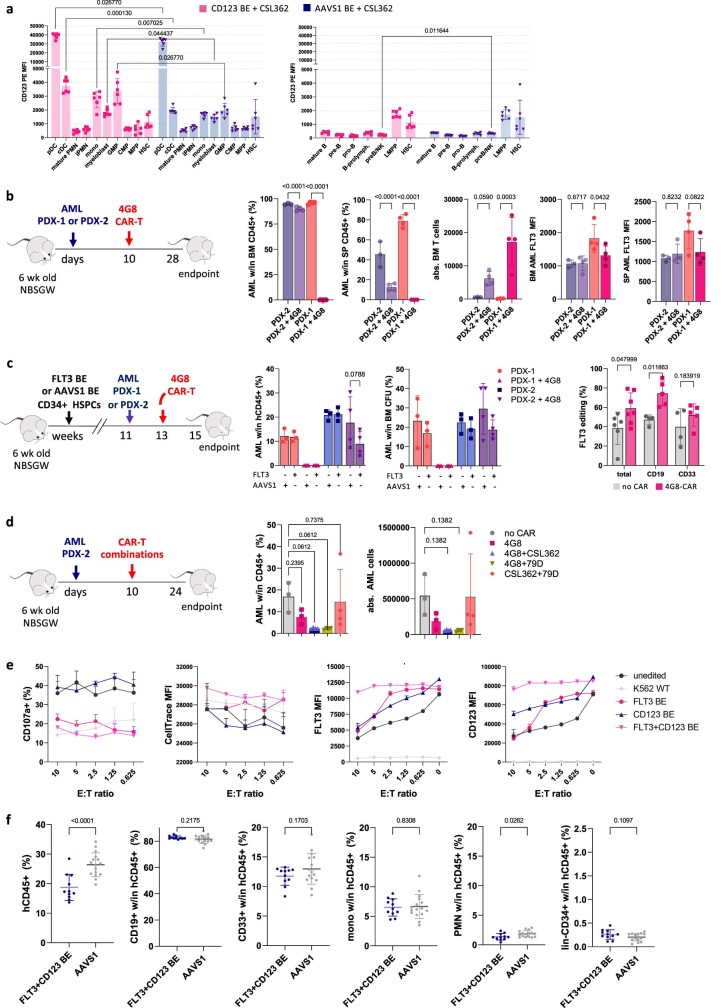
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
