

The microbiota conditions a gut milieu that selects for wild-type Salmonella Typhimurium virulence
- PMID: 37651408
- PMCID: PMC10499267
- DOI: 10.1371/journal.pbio.3002253
The microbiota conditions a gut milieu that selects for wild-type Salmonella Typhimurium virulence
Abstract
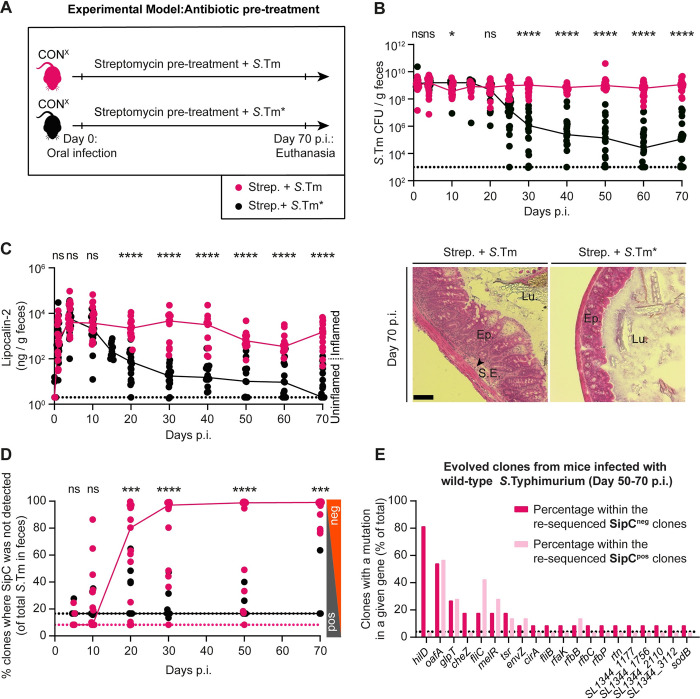
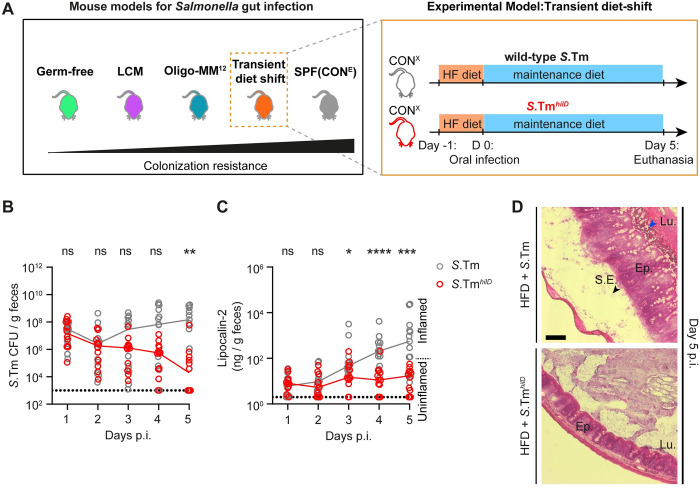
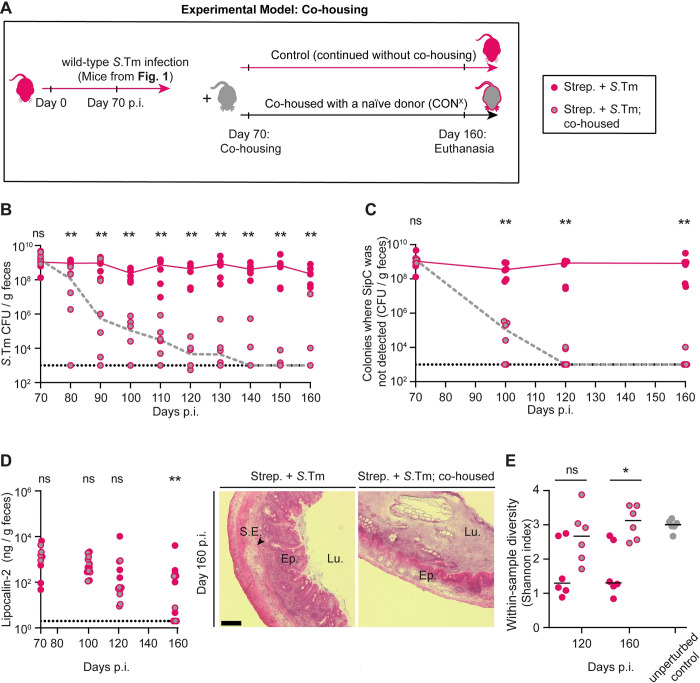
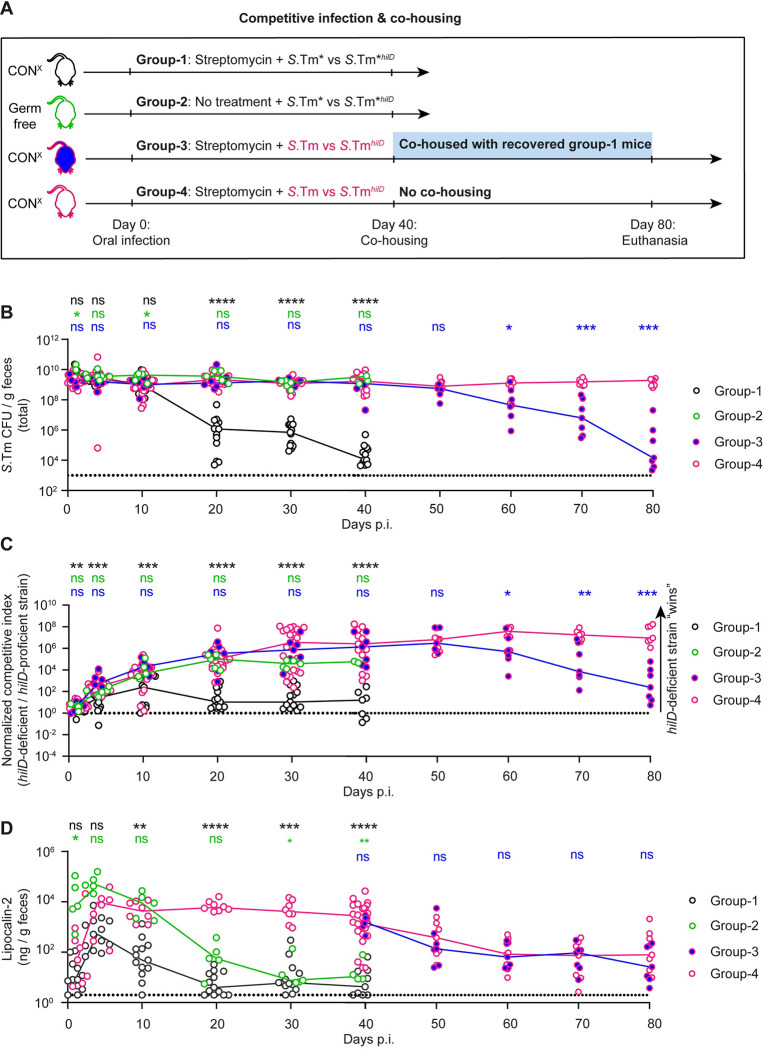
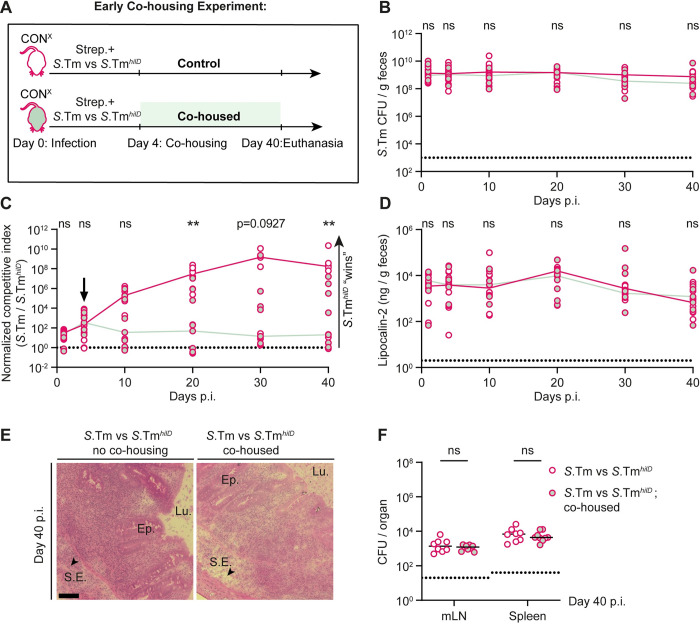
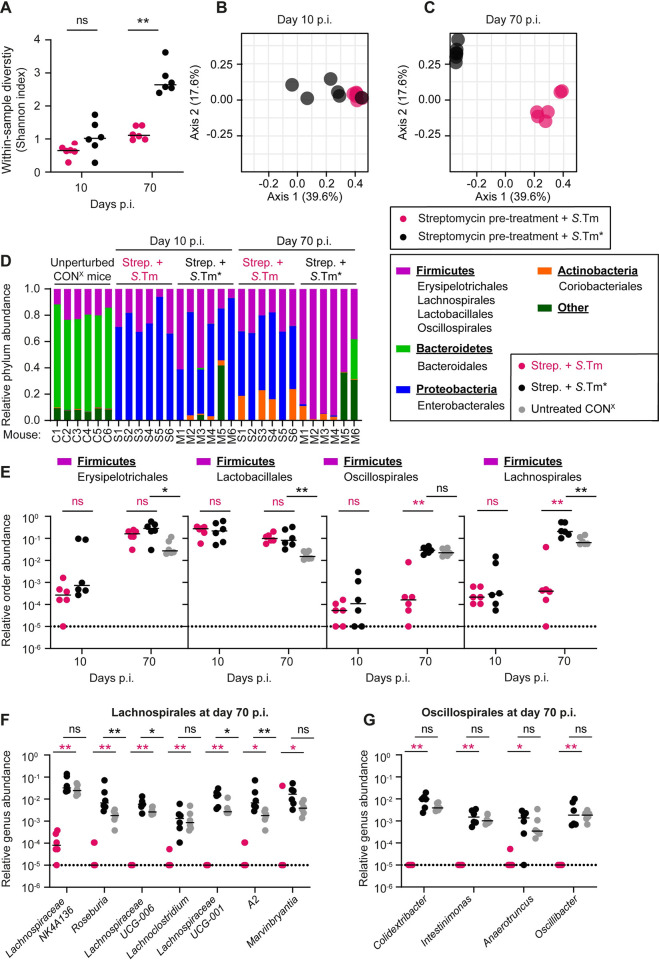
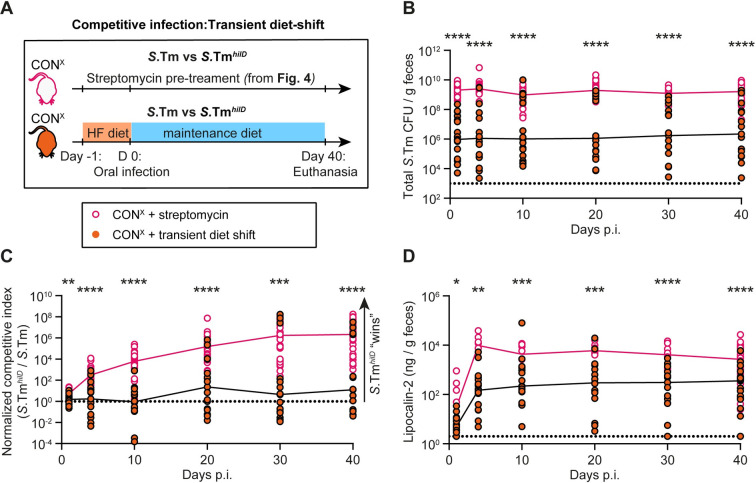
Salmonella Typhimurium elicits gut inflammation by the costly expression of HilD-controlled virulence factors. This inflammation alleviates colonization resistance (CR) mediated by the microbiota and thereby promotes pathogen blooms. However, the inflamed gut-milieu can also select for hilD mutants, which cannot elicit or maintain inflammation, therefore causing a loss of the pathogen's virulence. This raises the question of which conditions support the maintenance of virulence in S. Typhimurium. Indeed, it remains unclear why the wild-type hilD allele is dominant among natural isolates. Here, we show that microbiota transfer from uninfected or recovered hosts leads to rapid clearance of hilD mutants that feature attenuated virulence, and thereby contributes to the preservation of the virulent S. Typhimurium genotype. Using mouse models featuring a range of microbiota compositions and antibiotic- or inflammation-inflicted microbiota disruptions, we found that irreversible disruption of the microbiota leads to the accumulation of hilD mutants. In contrast, in models with a transient microbiota disruption, selection for hilD mutants was prevented by the regrowing microbiota community dominated by Lachnospirales and Oscillospirales. Strikingly, even after an irreversible microbiota disruption, microbiota transfer from uninfected donors prevented the rise of hilD mutants. Our results establish that robust S. Typhimurium gut colonization hinges on optimizing its manipulation of the host: A transient and tempered microbiota perturbation is favorable for the pathogen to both flourish in the inflamed gut and also minimize loss of virulence. Moreover, besides conferring CR, the microbiota may have the additional consequence of maintaining costly enteropathogen virulence mechanisms.
Copyright: © 2023 Gül et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Stabilization of cooperative virulence by the expression of an avirulent phenotype.Nature. 2013 Feb 21;494(7437):353-6. doi: 10.1038/nature11913. Nature. 2013. PMID: 23426324
-
Impact of horizontal gene transfer on emergence and stability of cooperative virulence in Salmonella Typhimurium.Nat Commun. 2022 Apr 11;13(1):1939. doi: 10.1038/s41467-022-29597-7. Nat Commun. 2022. PMID: 35410999 Free PMC article.
-
An incoherent feedforward loop formed by SirA/BarA, HilE and HilD is involved in controlling the growth cost of virulence factor expression by Salmonella Typhimurium.PLoS Pathog. 2021 May 28;17(5):e1009630. doi: 10.1371/journal.ppat.1009630. eCollection 2021 May. PLoS Pathog. 2021. PMID: 34048498 Free PMC article.
-
The streptomycin mouse model for Salmonella diarrhea: functional analysis of the microbiota, the pathogen's virulence factors, and the host's mucosal immune response.Immunol Rev. 2012 Jan;245(1):56-83. doi: 10.1111/j.1600-065X.2011.01070.x. Immunol Rev. 2012. PMID: 22168414 Review.
-
Salmonella Typhimurium Diarrhea Reveals Basic Principles of Enteropathogen Infection and Disease-Promoted DNA Exchange.Cell Host Microbe. 2017 Apr 12;21(4):443-454. doi: 10.1016/j.chom.2017.03.009. Cell Host Microbe. 2017. PMID: 28407482 Review.
Cited by
-
Emerging Strategies against Non-Typhoidal Salmonella: From Pathogenesis to Treatment.Curr Issues Mol Biol. 2024 Jul 14;46(7):7447-7472. doi: 10.3390/cimb46070442. Curr Issues Mol Biol. 2024. PMID: 39057083 Free PMC article. Review.
-
Attenuated Salmonella typhimurium L forms suppress tumor growth and promote apoptosis in murine ovarian tumors.Sci Rep. 2024 Jul 11;14(1):16045. doi: 10.1038/s41598-024-66898-x. Sci Rep. 2024. PMID: 38992056 Free PMC article.
-
Cannabinoid receptor deficiencies drive immune response dynamics in Salmonella infection.bioRxiv [Preprint]. 2025 Mar 11:2025.03.10.642352. doi: 10.1101/2025.03.10.642352. bioRxiv. 2025. PMID: 40161677 Free PMC article. Preprint.
-
Commensal E. coli limits Salmonella gut invasion during inflammation by producing toxin-bound siderophores in a tonB-dependent manner.PLoS Biol. 2024 Jun 12;22(6):e3002616. doi: 10.1371/journal.pbio.3002616. eCollection 2024 Jun. PLoS Biol. 2024. PMID: 38865418 Free PMC article.
-
Effect of diet-induced low-grade, chronic inflammation on susceptibility of broilers to Salmonella colonization and pathogenicity.Poult Sci. 2025 Aug;104(8):105303. doi: 10.1016/j.psj.2025.105303. Epub 2025 May 21. Poult Sci. 2025. PMID: 40480134 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
