

Epitope base editing CD45 in hematopoietic cells enables universal blood cancer immune therapy
- PMID: 37651540
- PMCID: PMC10682510
- DOI: 10.1126/scitranslmed.adi1145
Epitope base editing CD45 in hematopoietic cells enables universal blood cancer immune therapy
Abstract
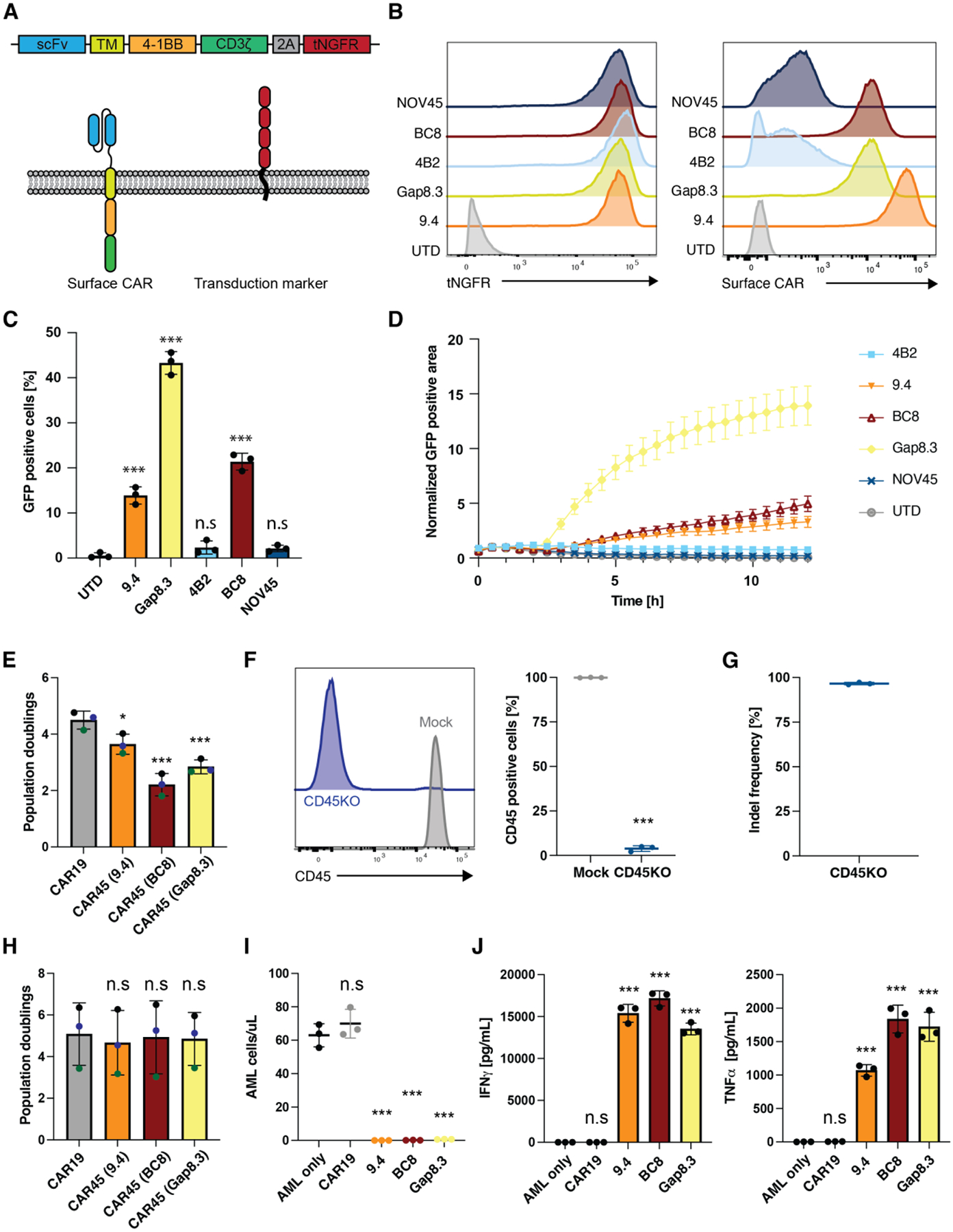
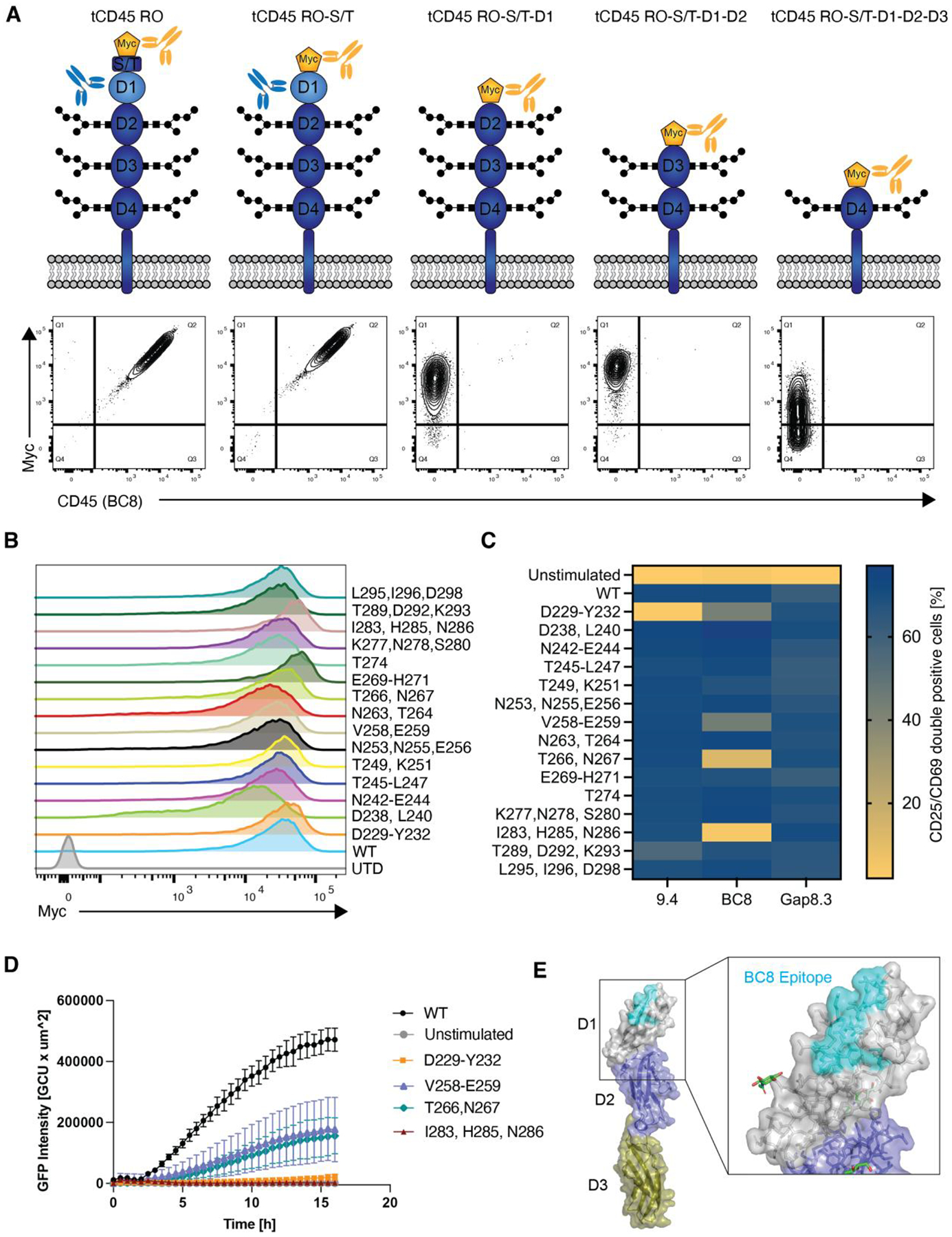
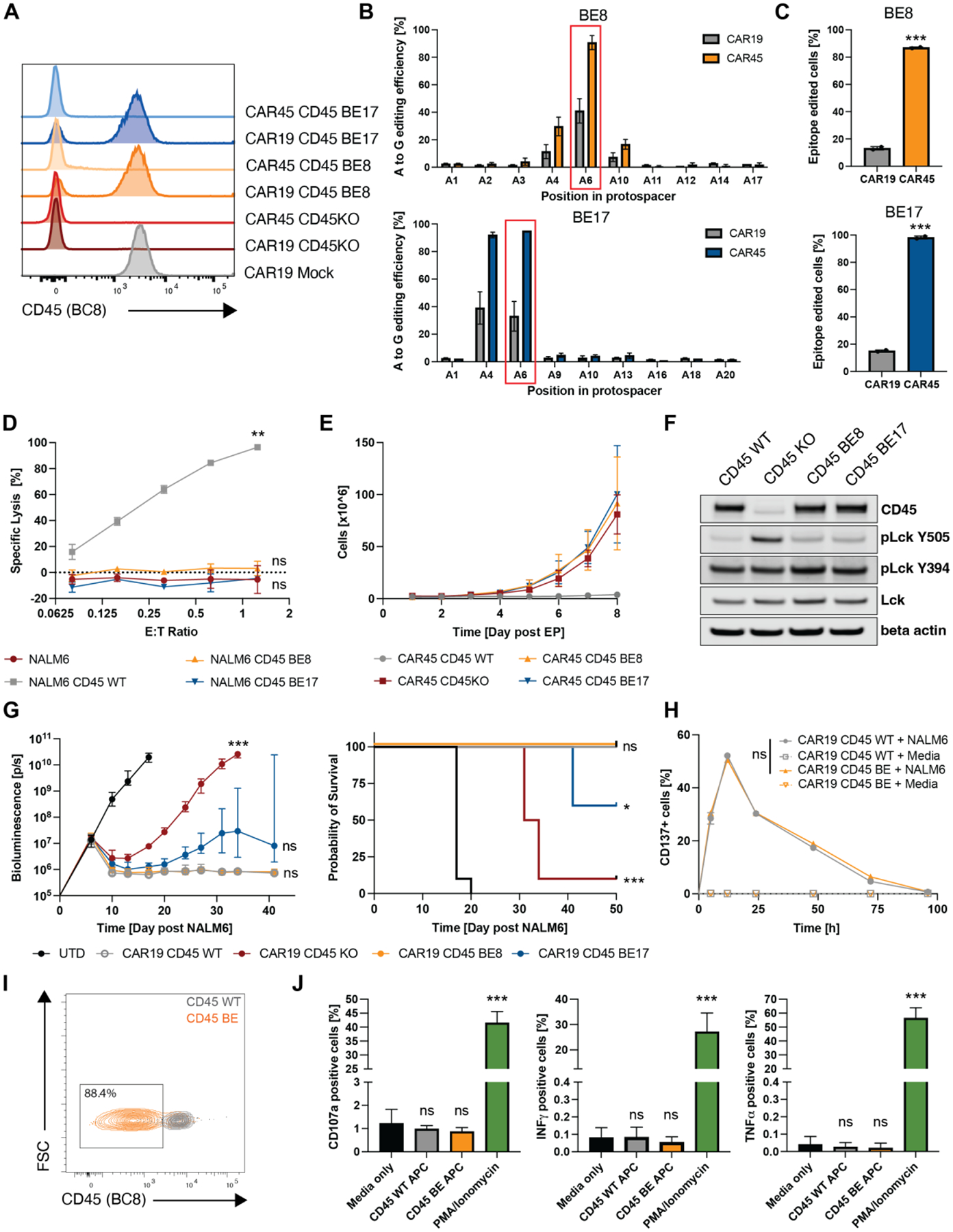
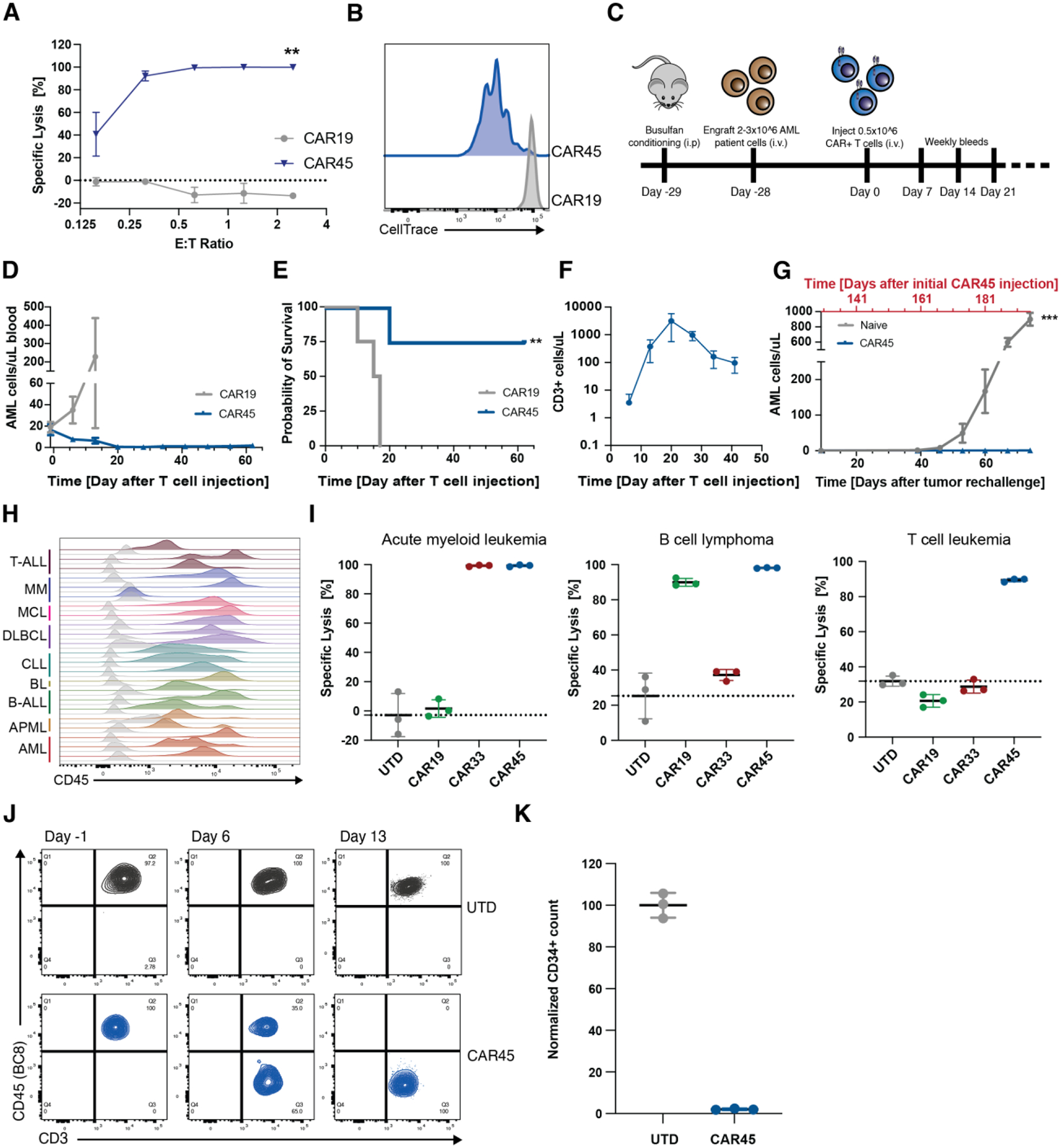
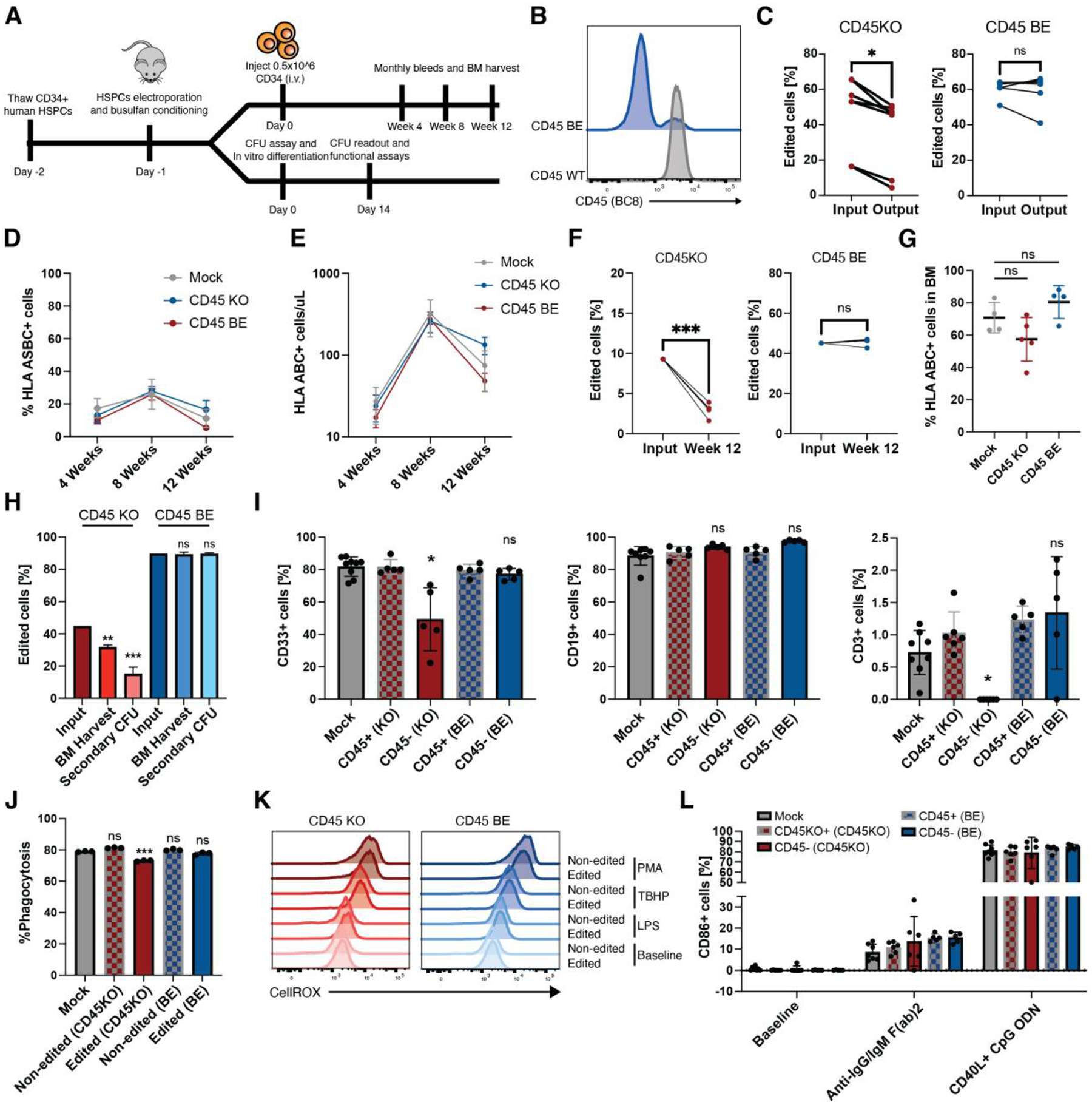
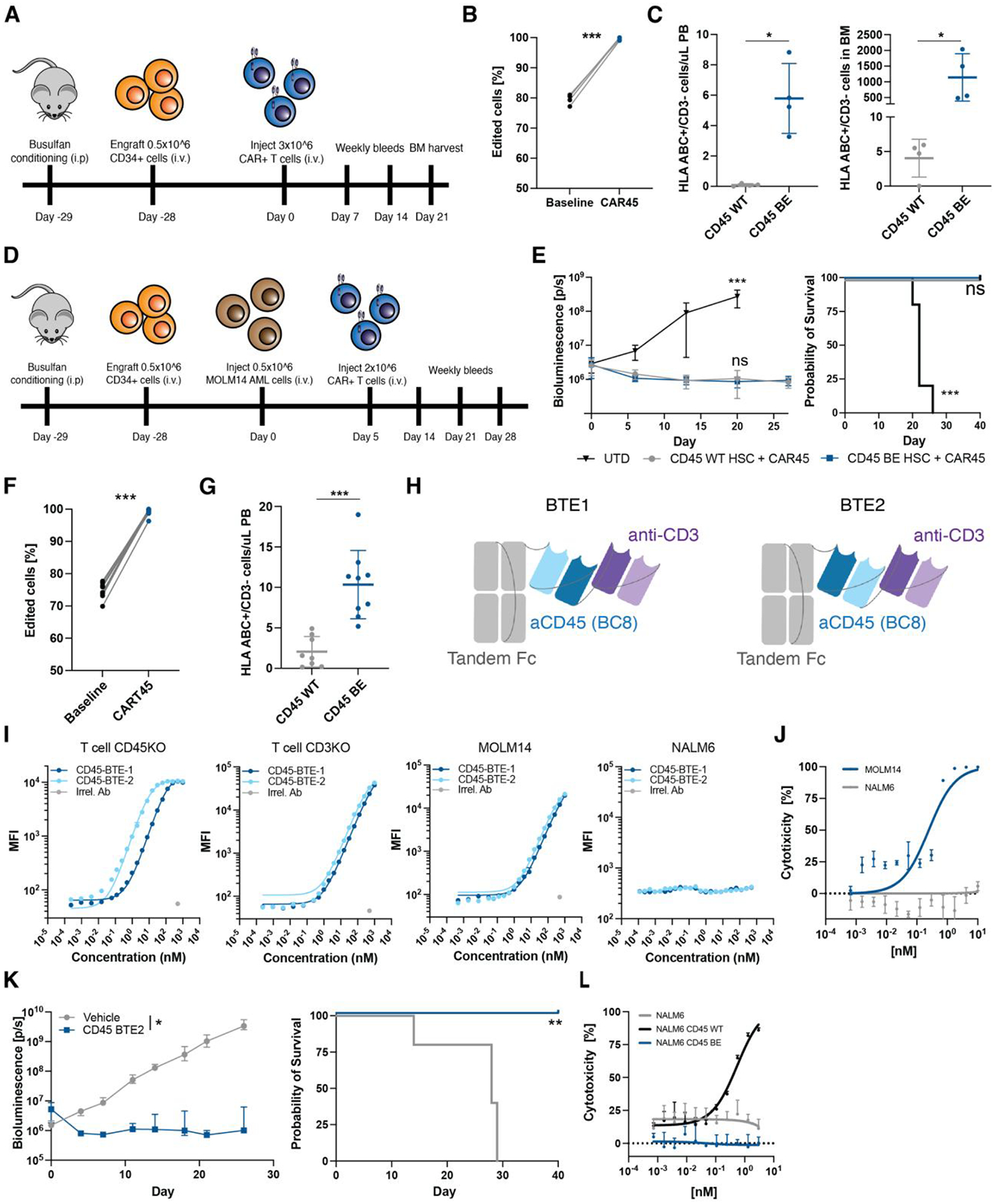
In the absence of cell surface cancer-specific antigens, immunotherapies such as chimeric antigen receptor (CAR) T cells, monoclonal antibodies, or bispecific T cell engagers typically target lineage antigens. Currently, such immunotherapies are individually designed and tested for each disease. This approach is inefficient and limited to a few lineage antigens for which the on-target/off-tumor toxicities are clinically tolerated. Here, we sought to develop a universal CAR T cell therapy for blood cancers directed against the pan-leukocyte marker CD45. To protect healthy hematopoietic cells, including CAR T cells, from CD45-directed on-target/off-tumor toxicity while preserving the essential functions of CD45, we mapped the epitope on CD45 that is targeted by the CAR and used CRISPR adenine base editing to install a function-preserving mutation sufficient to evade CAR T cell recognition. Epitope-edited CD45 CAR T cells were fratricide resistant and effective against patient-derived acute myeloid leukemia, B cell lymphoma, and acute T cell leukemia. Epitope-edited hematopoietic stem cells (HSCs) were protected from CAR T cells and, unlike CD45 knockout cells, could engraft, persist, and differentiate in vivo. Ex vivo epitope editing in HSCs and T cells enables the safe and effective use of CD45-directed CAR T cells and bispecific T cell engagers for the universal treatment of hematologic malignancies and might be exploited for other diseases requiring intensive hematopoietic ablation.
Conflict of interest statement
Authors (NW, KCG, CHJ, SIG) have filed an invention disclosure with the University of Pennsylvania and Stanford University based on this work.
C.H.J. has patents related to CAR therapy with royalties paid from Novartis to the University of Pennsylvania. C.H.J. is a scientific co-founder and holds equity in Capstan Therapeutics and Tmunity Therapeutics. C.H.J. serves on the board of AC Immune and is a scientific advisor to Alaunos, BluesphereBio, Cabaletta, Carisma, Cartography, Cellares, Cellcarta, Celldex, Danaher, Decheng, ImmuneSensor, Kite, Poseida, Verismo, Viracta, and WIRB-Copernicus group.
S.G. has patents related to CAR therapy with royalties paid from Novartis to the University of Pennsylvania. S.G. is a scientific co-founder and holds equity in Interius Biotherapeutics and Carisma Therapeutics. S.G. is a scientific advisor to Carisma, Cartography, Currus, Interius, Kite, NKILT, Mission Bio, and Vor Bio.
DW has received grant funding from industry for sponsored research collaborations, he has received speaking honoraria, and received fees for consulting or serving on scientific review committees. Remunerations received by DW include direct payments and equity/options. DW also discloses the following associations with commercial partners: Geneos consultant/advisory board, AstraZeneca advisory board, speaker, Inovio board of directors, consultant, Sanofi advisory board, BBI advisory board, Pfizer advisory Board, Flagship consultant, and Advaccine consultant.
R.M.Y. is an inventor on patents and/or patent applications licensed to Novartis Institutes of Biomedical Research and Tmunity Therapeutics and receives license revenue from such licenses.
Figures
References
-
- Zammarchi F et al. , ADCT-402, a PBD dimer–containing antibody drug conjugate targeting CD19-expressing malignancies. Blood, The Journal of the American Society of Hematology 131, 1094–1105 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
