

Pathogen-driven CRISPR screens identify TREX1 as a regulator of DNA self-sensing during influenza virus infection
- PMID: 37652009
- PMCID: PMC10528757
- DOI: 10.1016/j.chom.2023.08.001
Pathogen-driven CRISPR screens identify TREX1 as a regulator of DNA self-sensing during influenza virus infection
Abstract
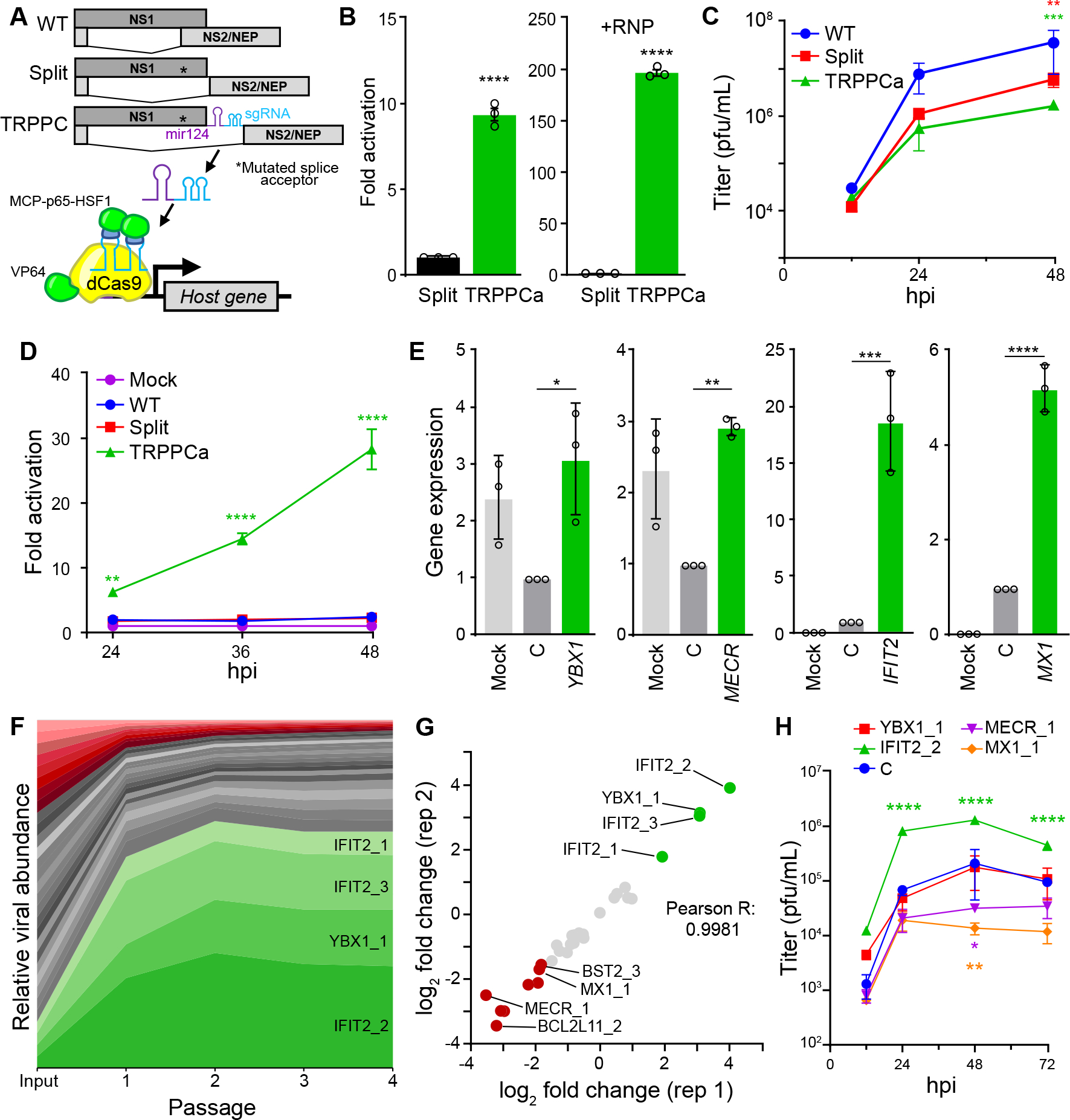
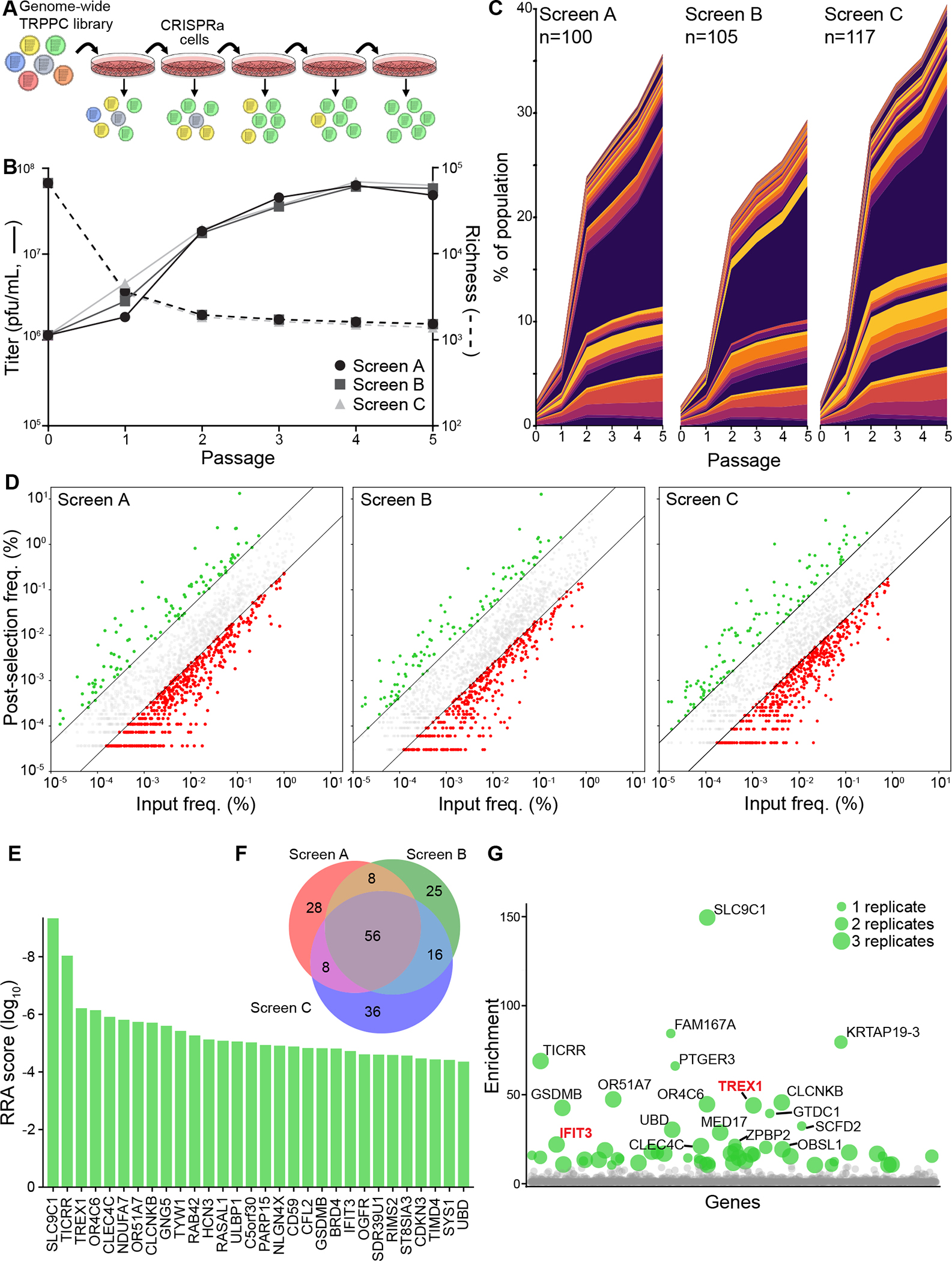
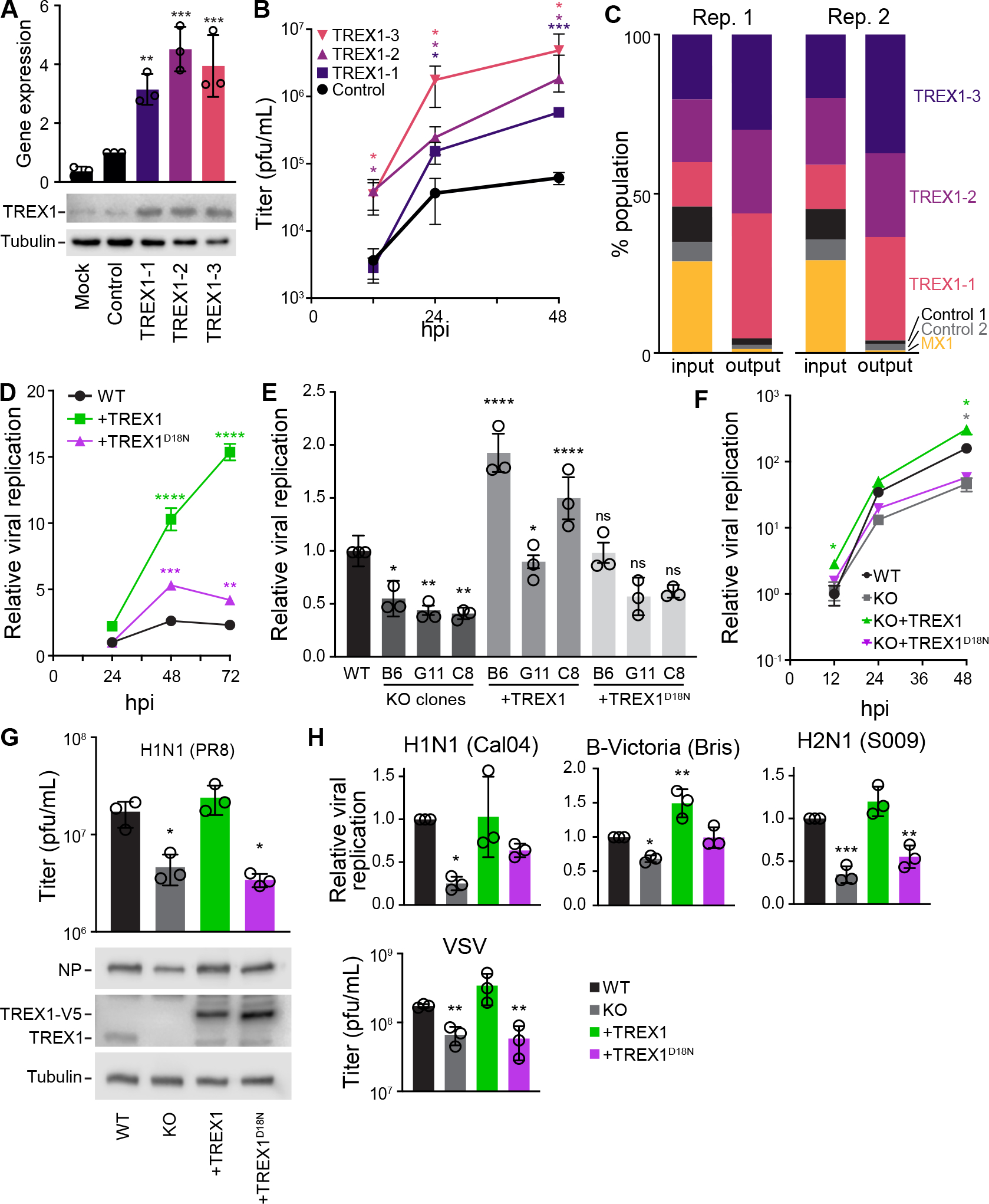
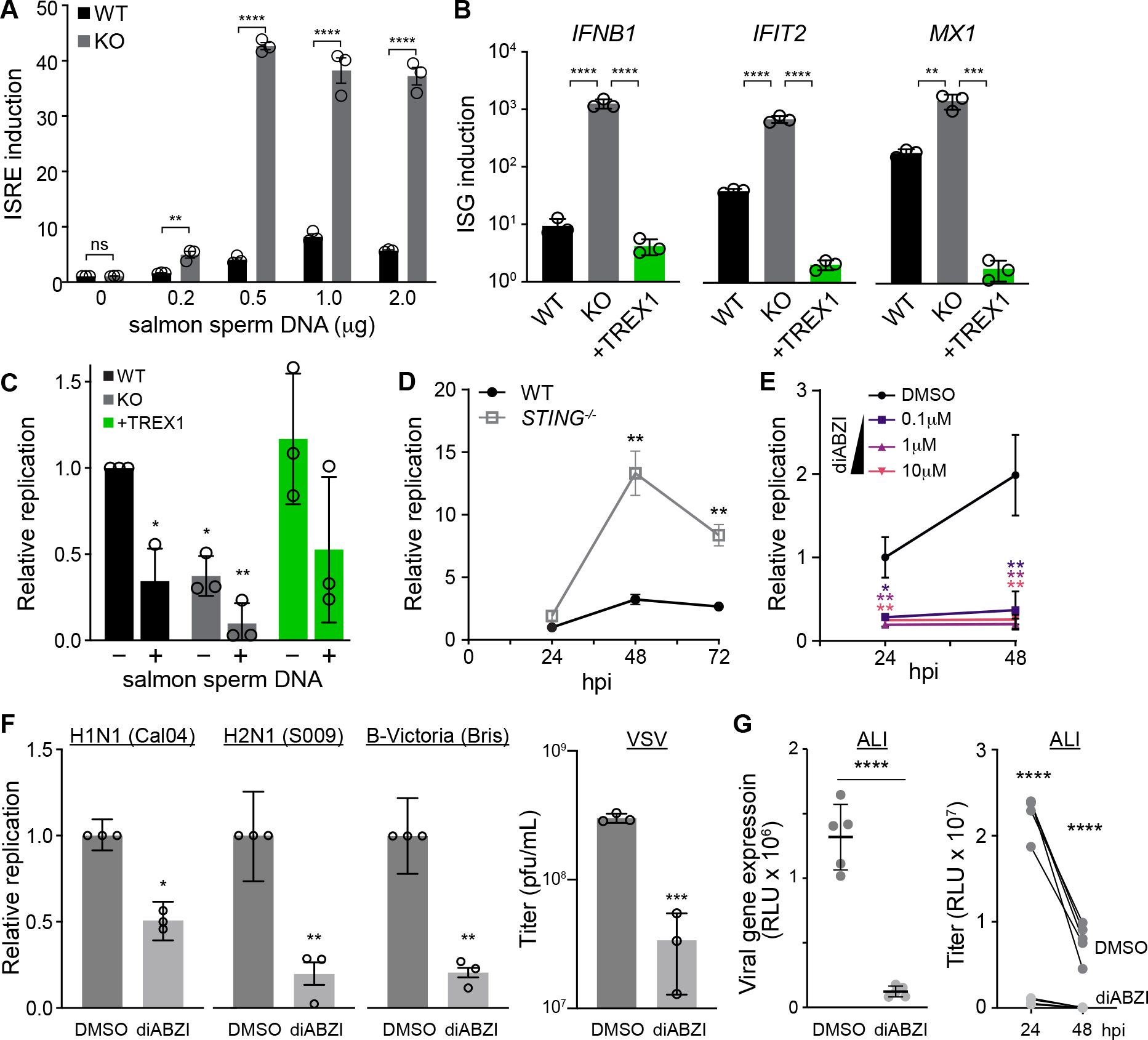
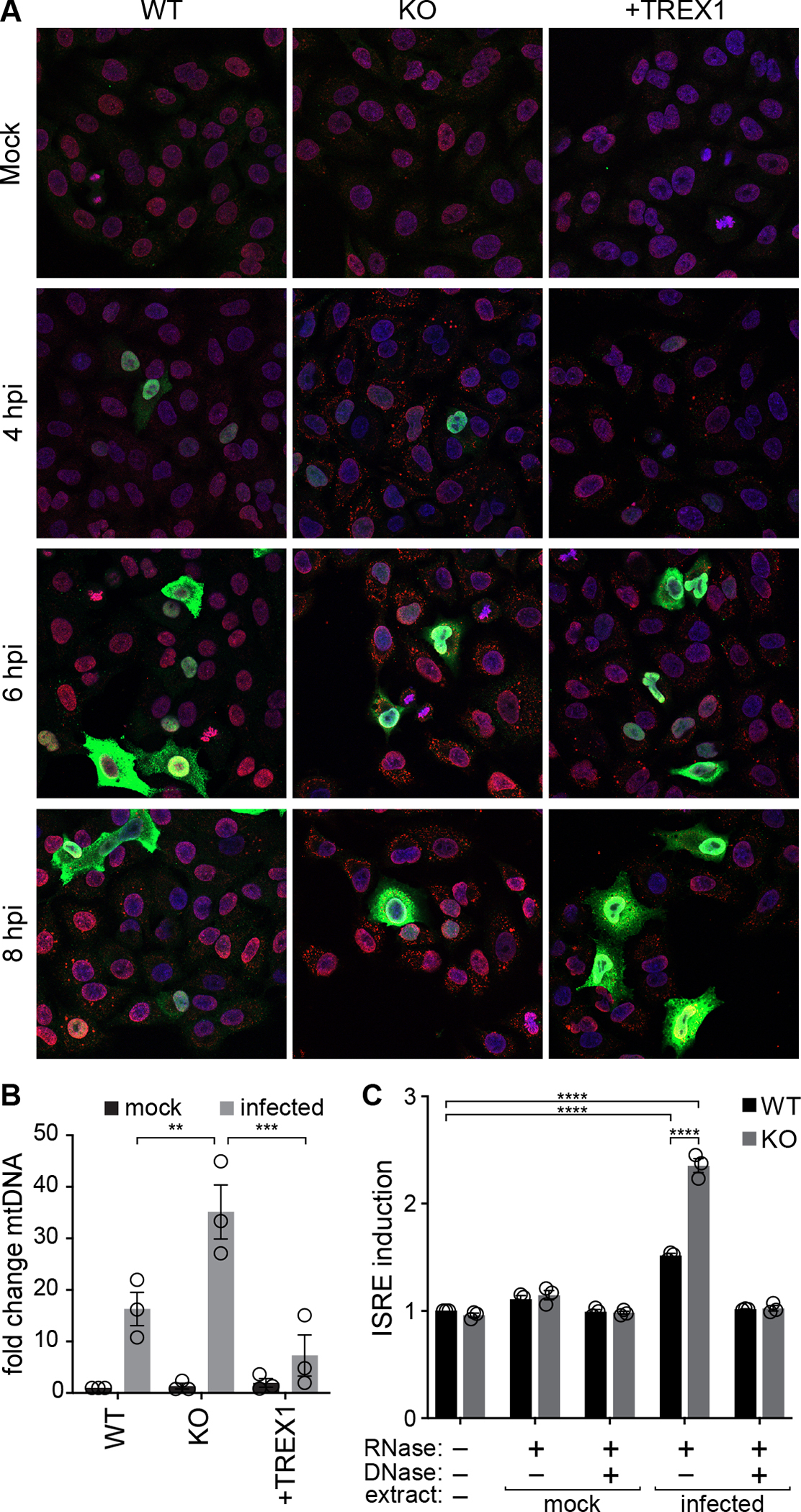
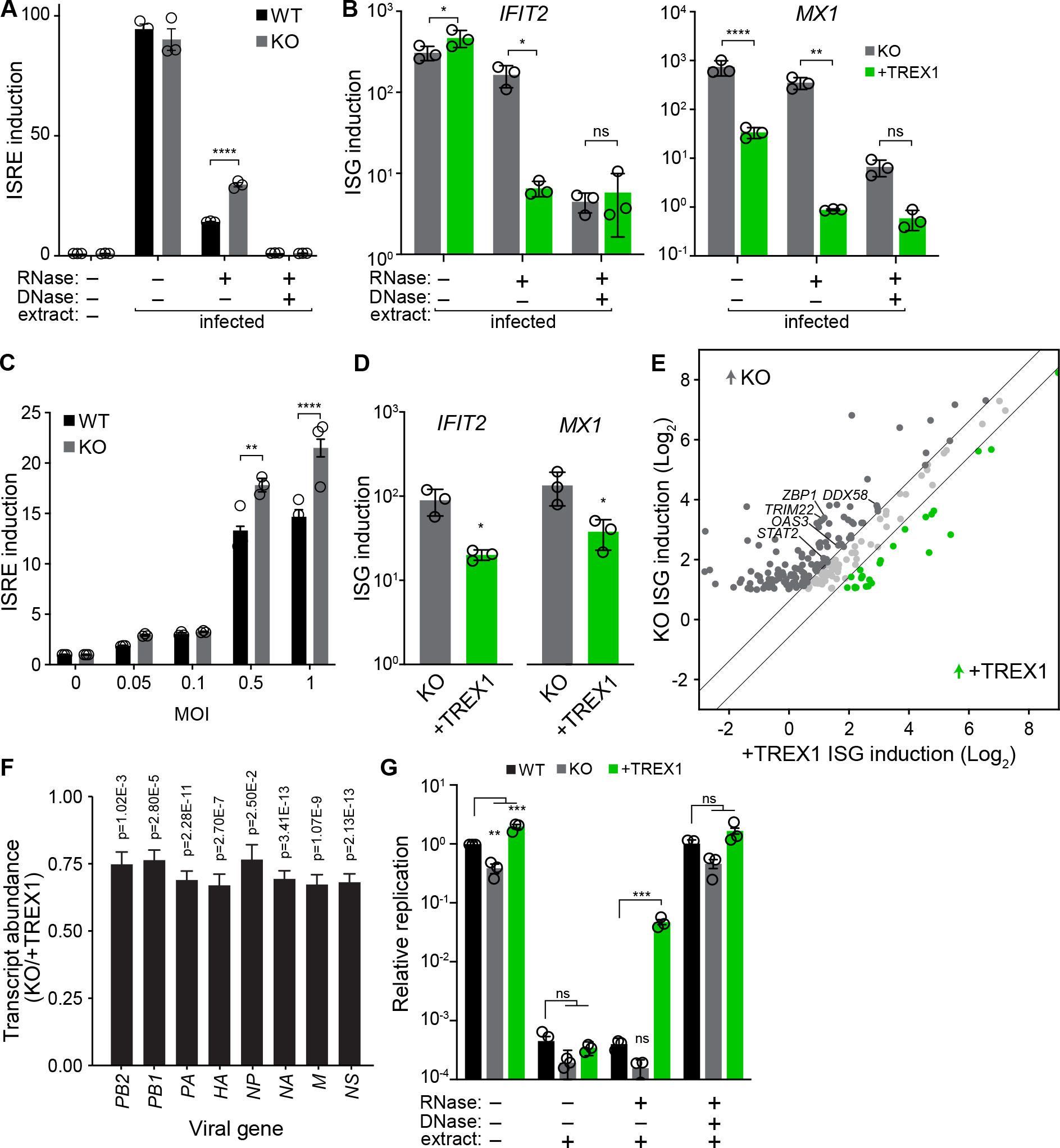
Host:pathogen interactions dictate the outcome of infection, yet the limitations of current approaches leave large regions of this interface unexplored. Here, we develop a novel fitness-based screen that queries factors important during the middle to late stages of infection. This is achieved by engineering influenza virus to direct the screen by programming dCas9 to modulate host gene expression. Our genome-wide screen for pro-viral factors identifies the cytoplasmic DNA exonuclease TREX1. TREX1 degrades cytoplasmic DNA to prevent inappropriate innate immune activation by self-DNA. We reveal that this same process aids influenza virus replication. Infection triggers release of mitochondrial DNA into the cytoplasm, activating antiviral signaling via cGAS and STING. TREX1 metabolizes the DNA, preventing its sensing. Collectively, these data show that self-DNA is deployed to amplify innate immunity, a process tempered by TREX1. Moreover, they demonstrate the power and generality of pathogen-driven fitness-based screens to pinpoint key host regulators of infection.
Keywords: CRISPR activation; TREX1; fitness-based screen; influenza virus; mtDNA.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests C.R.K. and A.M. are inventors on a provisional patent related to this work.
Figures
Update of
-
Pathogen-driven CRISPR screens identify TREX1 as a regulator of DNA self-sensing during influenza virus infection.bioRxiv [Preprint]. 2023 Feb 7:2023.02.07.527556. doi: 10.1101/2023.02.07.527556. bioRxiv. 2023. Update in: Cell Host Microbe. 2023 Sep 13;31(9):1552-1567.e8. doi: 10.1016/j.chom.2023.08.001. PMID: 36798235 Free PMC article. Updated. Preprint.
References
-
- Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, et al. (2009). The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139, 1243–1254. 10.1016/j.cell.2009.12.017. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
