

The extracellular matrix in hepatocellular carcinoma: Mechanisms and therapeutic vulnerability
- PMID: 37652015
- PMCID: PMC10518608
- DOI: 10.1016/j.xcrm.2023.101170
The extracellular matrix in hepatocellular carcinoma: Mechanisms and therapeutic vulnerability
Abstract
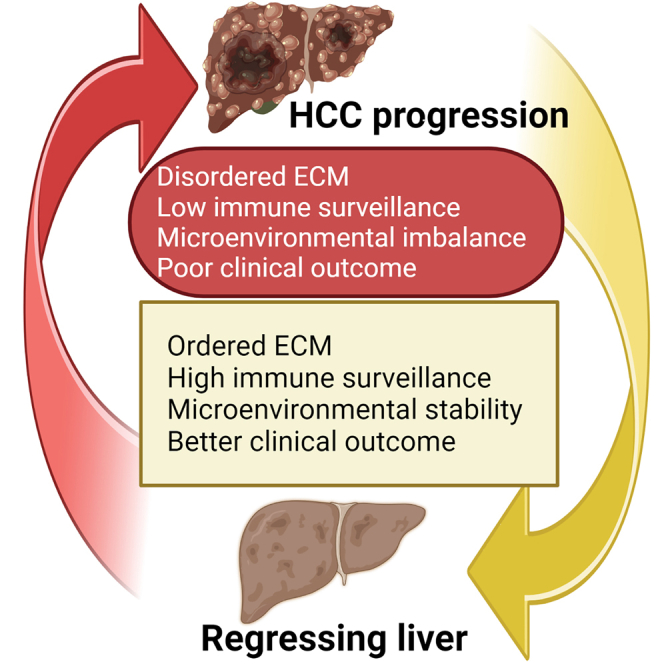
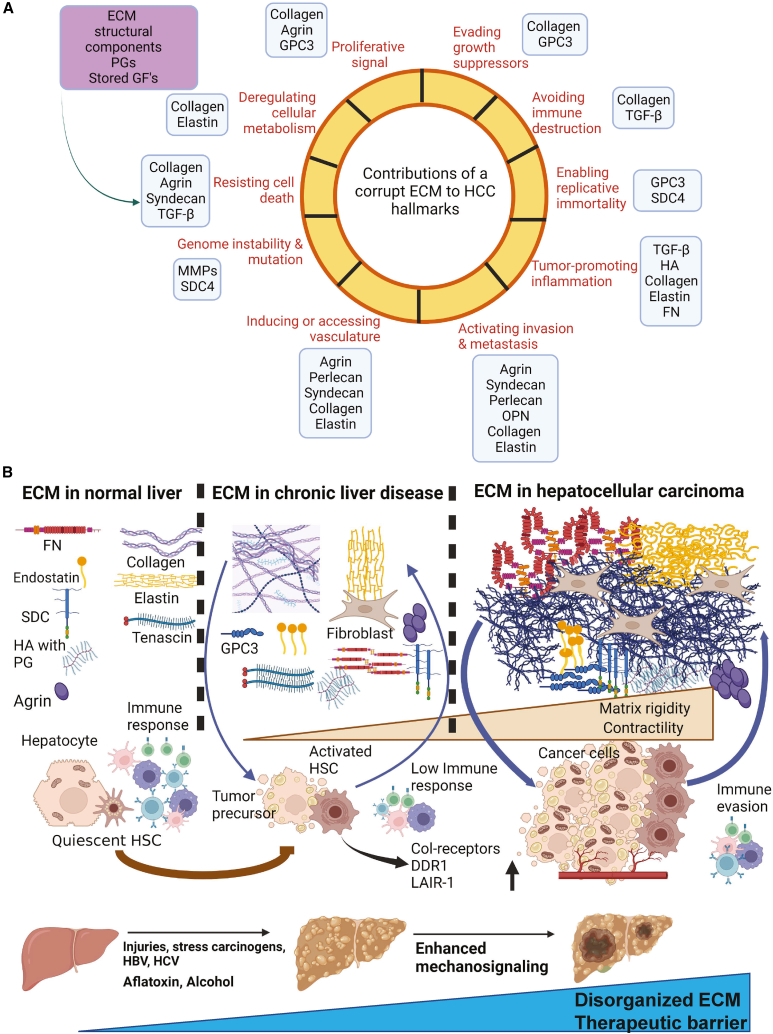
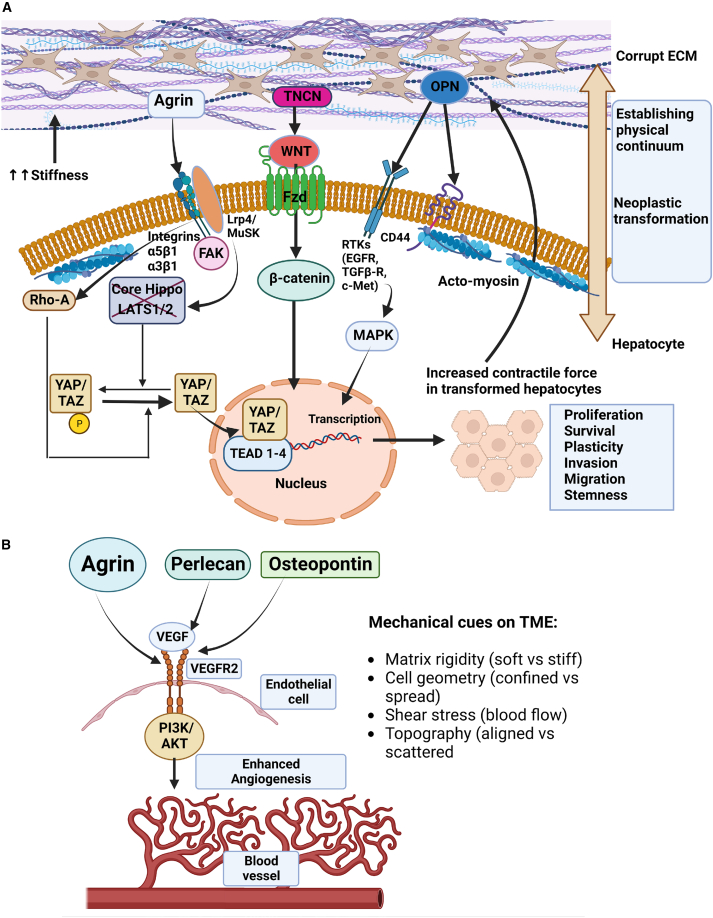
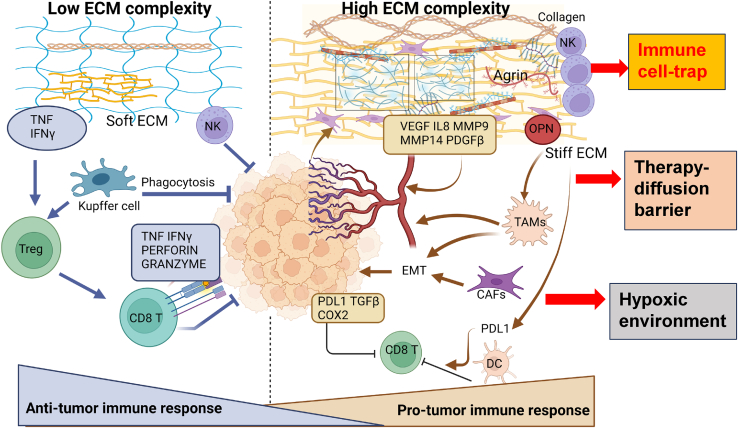
The tumor microenvironment (TME) is influenced by a "disorganized" extracellular matrix (ECM) that sensitizes cancer cells toward mechanical stress, signaling, and structural alterations. In hepatocellular carcinoma (HCC), lack of knowledge about key ECM proteins driving the TME refractory to targeted therapies poses a barrier to the identification of new therapeutic targets. Herein, we discuss the contributions of various ECM components that impact hepatocytes and their surrounding support network during tumorigenesis. In addition, the underpinnings by which ECM proteins transduce mechanical signals to the liver TME are detailed. Finally, in view of the bidirectional feedback between the ECM, transformed hepatocytes, and immune cells, we highlight the potential role of the ECM disorganization process in shaping responses to immune checkpoint inhibitors and targeted therapies. Our comprehensive characterization of these ECM components may provide a roadmap for innovative therapeutic approaches to restrain HCC.
Keywords: YAP/TAZ; agrin; chronic liver inflammation; collagen; extracellular matrix; fibrosis; glypican-3; hepatocellular carcinoma; immunotherapy; mechanotransduction; targeted therapies.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Similar articles
-
Unveiling the Role of Mechanical Microenvironment in Hepatocellular Carcinoma: Molecular Mechanisms and Implications for Therapeutic Strategies.Int J Biol Sci. 2024 Sep 30;20(13):5239-5253. doi: 10.7150/ijbs.102706. eCollection 2024. Int J Biol Sci. 2024. PMID: 39430235 Free PMC article. Review.
-
Agrin Mediates Angiogenesis in the Tumor Microenvironment.Trends Cancer. 2020 Feb;6(2):81-85. doi: 10.1016/j.trecan.2019.12.002. Epub 2020 Jan 9. Trends Cancer. 2020. PMID: 32061308
-
Regulation of tumor immunity and immunotherapy by the tumor collagen extracellular matrix.Front Immunol. 2023 Aug 17;14:1199513. doi: 10.3389/fimmu.2023.1199513. eCollection 2023. Front Immunol. 2023. PMID: 37662958 Free PMC article. Review.
-
Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy.J Hepatol. 2023 Apr;78(4):770-782. doi: 10.1016/j.jhep.2023.01.011. Epub 2023 Jan 26. J Hepatol. 2023. PMID: 36708811
-
Tumor microenvironment-mediated immune evasion in hepatocellular carcinoma.Front Immunol. 2023 Feb 10;14:1133308. doi: 10.3389/fimmu.2023.1133308. eCollection 2023. Front Immunol. 2023. PMID: 36845131 Free PMC article. Review.
Cited by
-
Partial Hepatectomy Promotes the Development of KRASG12V-Induced Hepatocellular Carcinoma in Zebrafish.Cancers (Basel). 2024 May 8;16(10):1793. doi: 10.3390/cancers16101793. Cancers (Basel). 2024. PMID: 38791872 Free PMC article.
-
Tim-1-mediated extracellular matrix promotes the development of hepatocellular carcinoma.Acta Biochim Biophys Sin (Shanghai). 2024 Oct 23;56(12):1761-1773. doi: 10.3724/abbs.2024191. Acta Biochim Biophys Sin (Shanghai). 2024. PMID: 39444345 Free PMC article.
-
Sorafenib-associated translation reprogramming in hepatocellular carcinoma cells.RNA Biol. 2025 Dec;22(1):1-11. doi: 10.1080/15476286.2025.2483484. Epub 2025 Mar 24. RNA Biol. 2025. PMID: 40116042 Free PMC article.
-
An Agrin-YAP/TAZ Rigidity Sensing Module Drives EGFR-Addicted Lung Tumorigenesis.Adv Sci (Weinh). 2025 May;12(20):e2413443. doi: 10.1002/advs.202413443. Epub 2025 Mar 31. Adv Sci (Weinh). 2025. PMID: 40165020 Free PMC article.
-
Chitosan-based biomaterial delivery strategies for hepatocellular carcinoma.Front Pharmacol. 2024 Aug 5;15:1446030. doi: 10.3389/fphar.2024.1446030. eCollection 2024. Front Pharmacol. 2024. PMID: 39161903 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
