

The penetrance of rare variants in cardiomyopathy-associated genes: A cross-sectional approach to estimating penetrance for secondary findings
- PMID: 37652022
- PMCID: PMC10502871
- DOI: 10.1016/j.ajhg.2023.08.003
The penetrance of rare variants in cardiomyopathy-associated genes: A cross-sectional approach to estimating penetrance for secondary findings
Abstract
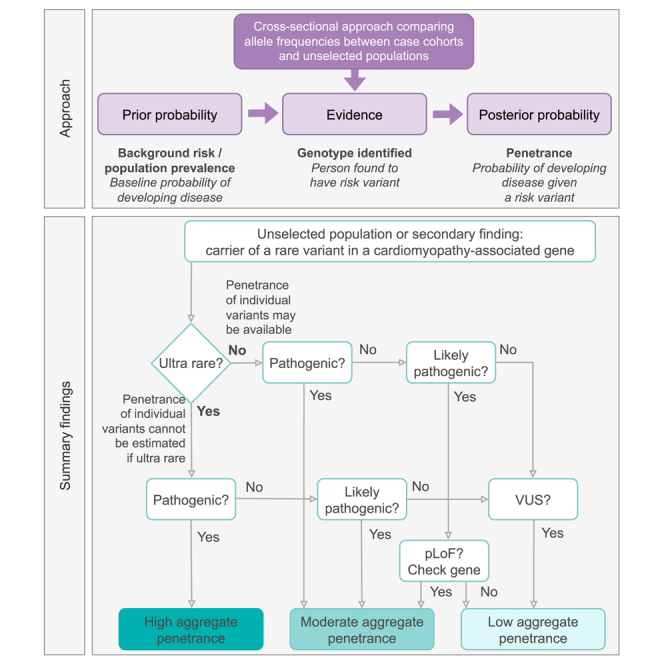
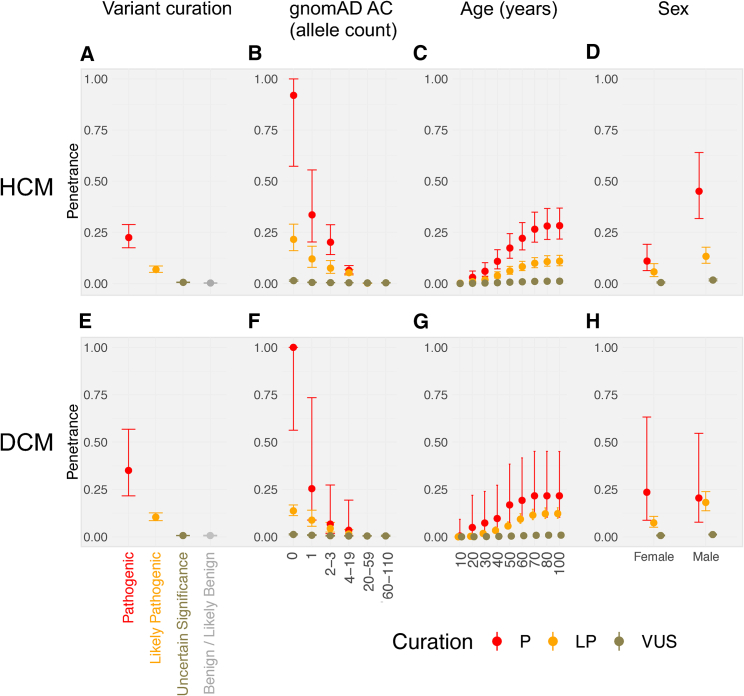
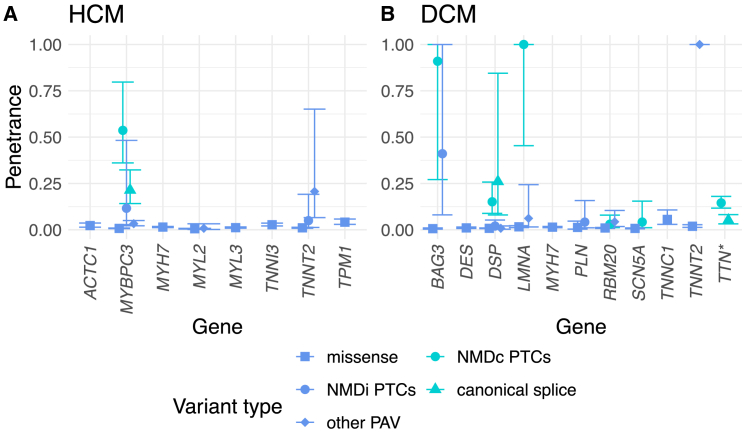
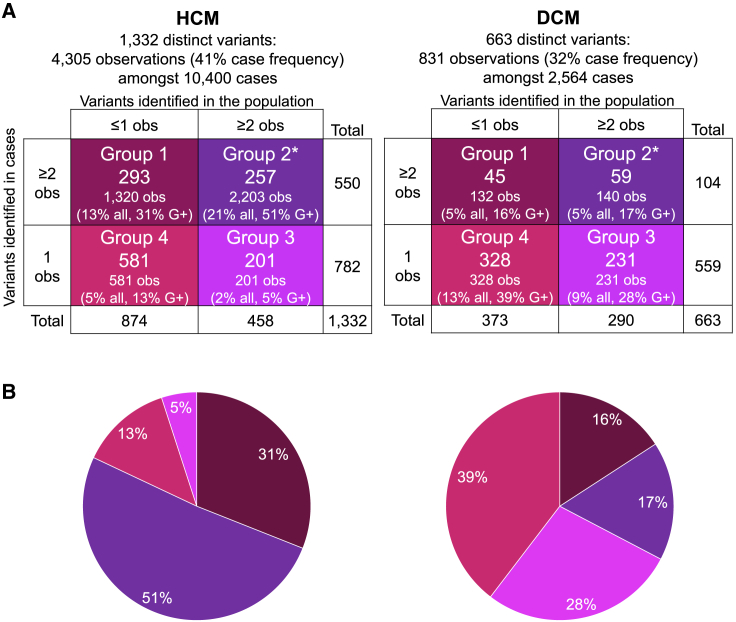
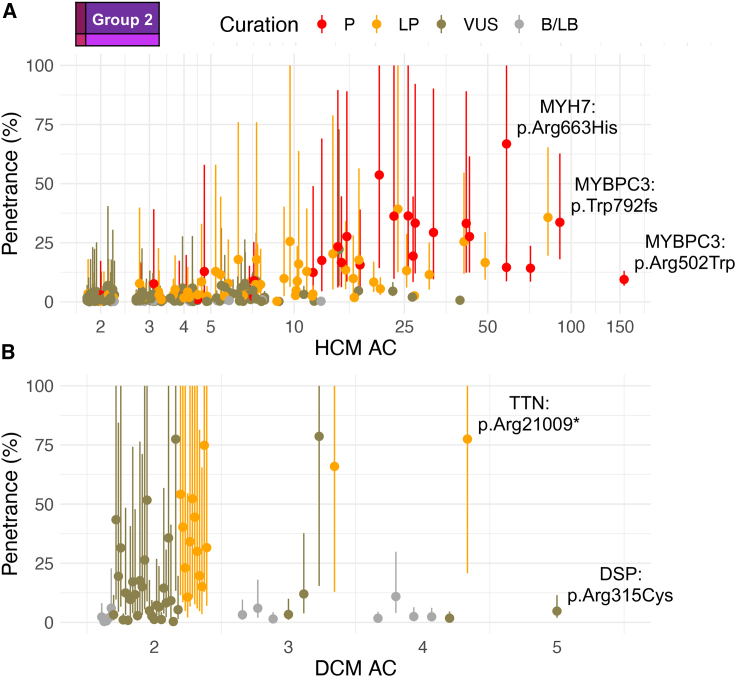
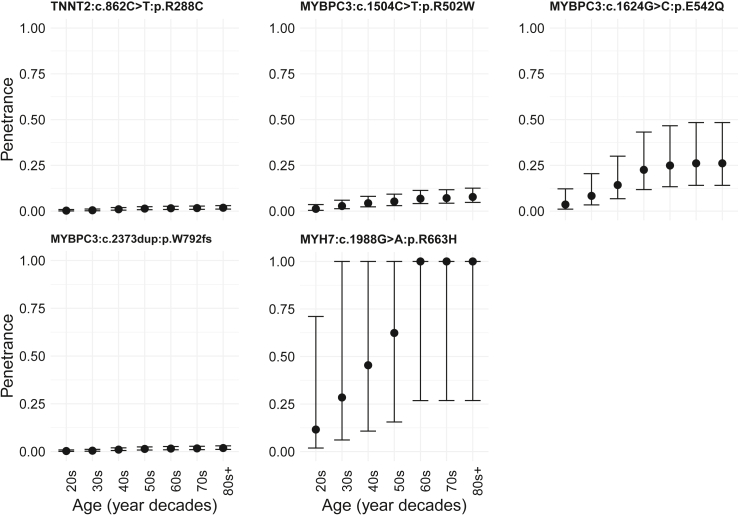
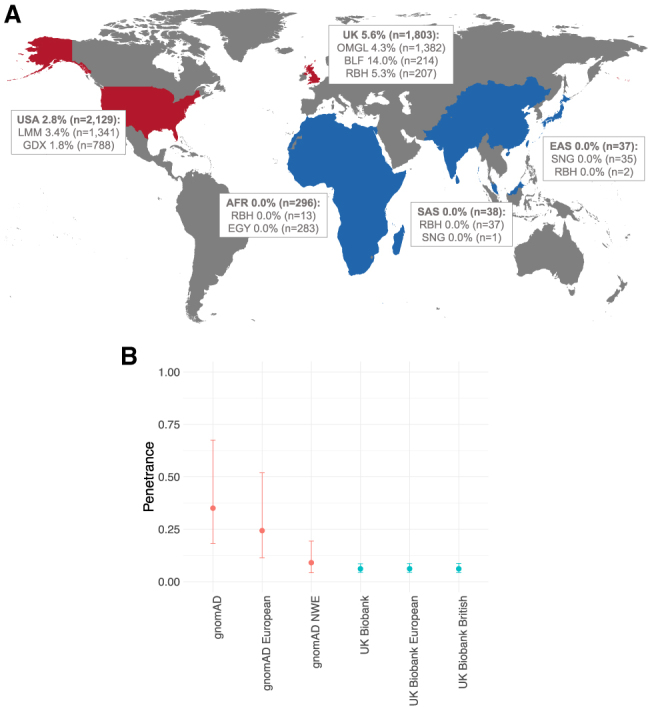
Understanding the penetrance of pathogenic variants identified as secondary findings (SFs) is of paramount importance with the growing availability of genetic testing. We estimated penetrance through large-scale analyses of individuals referred for diagnostic sequencing for hypertrophic cardiomyopathy (HCM; 10,400 affected individuals, 1,332 variants) and dilated cardiomyopathy (DCM; 2,564 affected individuals, 663 variants), using a cross-sectional approach comparing allele frequencies against reference populations (293,226 participants from UK Biobank and gnomAD). We generated updated prevalence estimates for HCM (1:543) and DCM (1:220). In aggregate, the penetrance by late adulthood of rare, pathogenic variants (23% for HCM, 35% for DCM) and likely pathogenic variants (7% for HCM, 10% for DCM) was substantial for dominant cardiomyopathy (CM). Penetrance was significantly higher for variant subgroups annotated as loss of function or ultra-rare and for males compared to females for variants in HCM-associated genes. We estimated variant-specific penetrance for 316 recurrent variants most likely to be identified as SFs (found in 51% of HCM- and 17% of DCM-affected individuals). 49 variants were observed at least ten times (14% of affected individuals) in HCM-associated genes. Median penetrance was 14.6% (±14.4% SD). We explore estimates of penetrance by age, sex, and ancestry and simulate the impact of including future cohorts. This dataset reports penetrance of individual variants at scale and will inform the management of individuals undergoing genetic screening for SFs. While most variants had low penetrance and the costs and harms of screening are unclear, some individuals with highly penetrant variants may benefit from SFs.
Keywords: cardiomyopathy; penetrance; prevalence; secondary findings.
Copyright © 2023. Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests J.S.W. has consulted for MyoKardia, Inc., Foresite Labs, and Pfizer. A.H. now works for AstraZeneca, UK. D.P.O. has consulted for Bayer. L.B. has consulted for Roche. D.G.M. is a paid advisor to GlaxoSmithKline, Insitro, Variant Bio, and Overtone Therapeutics and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Merck, Pfizer, and Sanofi-Genzyme; none of these activities are directly related to the work presented here. E.M. is the owner of Mazalytics LLC, Boston, Massachusetts, USA.
Figures
References
-
- Miller D.T., Lee K., Abul-Husn N.S., Amendola L.M., Brothers K., Chung W.K., Gollob M.H., Gordon A.S., Harrison S.M., Hershberger R.E., et al. ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG) Genet. Med. 2022;24:1407–1414. doi: 10.1016/j.gim.2022.04.006. - DOI - PubMed
-
- de Wert G., Dondorp W., Clarke A., Dequeker E.M.C., Cordier C., Deans Z., van El C.G., Fellmann F., Hastings R., Hentze S., et al. Opportunistic genomic screening. Recommendations of the European Society of Human Genetics. Eur. J. Hum. Genet. 2021;29:365–377. doi: 10.1038/s41431-020-00758-w. - DOI - PMC - PubMed
-
- Ormondroyd E., Mackley M.P., Blair E., Craft J., Knight J.C., Taylor J.C., Taylor J., Watkins H. “Not pathogenic until proven otherwise”: Perspectives of UK clinical genomics professionals toward secondary findings in context of a Genomic Medicine Multidisciplinary Team and the 100,000 Genomes Project. Genet. Med. 2018;20:320–328. doi: 10.1038/gim.2017.157. - DOI - PMC - PubMed
-
- McGurk K.A., Zheng S.L., Henry A., Josephs K., Edwards M., de Marvao A., Whiffin N., Roberts A., Lumbers T.R., O’Regan D.P., Ware J.S. Correspondence on "ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2022;24:744–746. doi: 10.1016/j.gim.2021.10.020. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
- MC_U120085815/MRC_/Medical Research Council/United Kingdom
- RG/18/9/33887/BHF_/British Heart Foundation/United Kingdom
- MC-A658-5TY00/MRC_/Medical Research Council/United Kingdom
- DH_/Department of Health/United Kingdom
- BBC/F/21/220106/BHF_/British Heart Foundation/United Kingdom
- FS/IPBSRF/22/27059/BHF_/British Heart Foundation/United Kingdom
- MC_UP_1605/13/MRC_/Medical Research Council/United Kingdom
- RG/19/6/34387/BHF_/British Heart Foundation/United Kingdom
- RE/18/4/34215/BHF_/British Heart Foundation/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- 200990/A/16/Z/WT_/Wellcome Trust/United Kingdom
- MC_UP_1102/20/MRC_/Medical Research Council/United Kingdom
- 107469/Z/15/Z/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
