

Profiling of repetitive RNA sequences in the blood plasma of patients with cancer
- PMID: 37652985
- PMCID: PMC10727983
- DOI: 10.1038/s41551-023-01081-7
Profiling of repetitive RNA sequences in the blood plasma of patients with cancer
Abstract
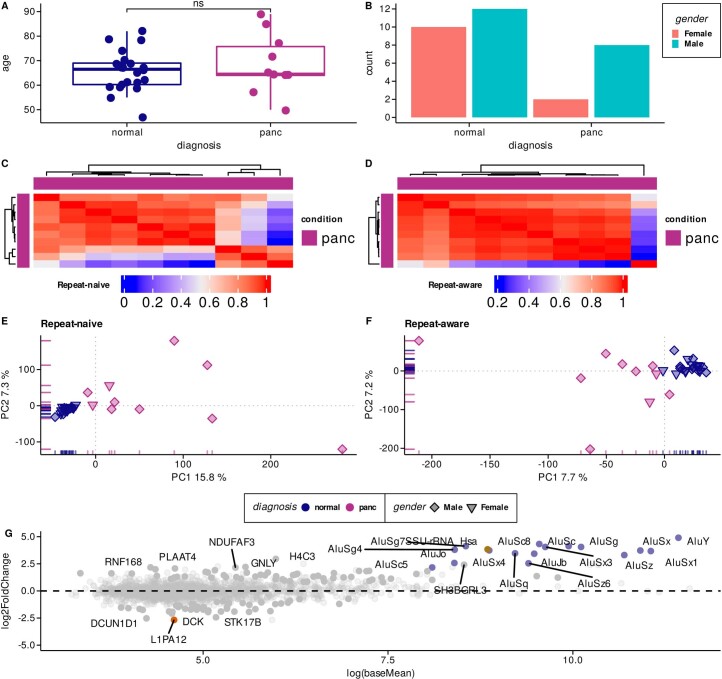
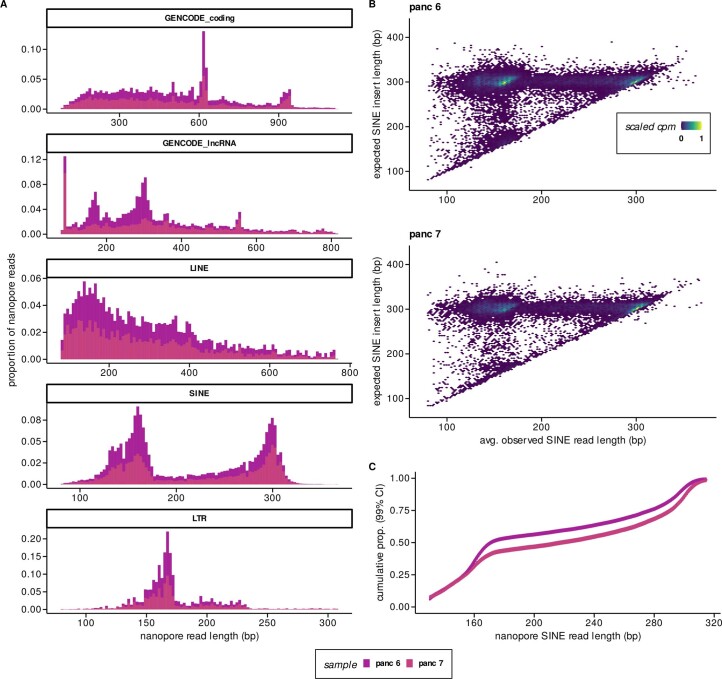
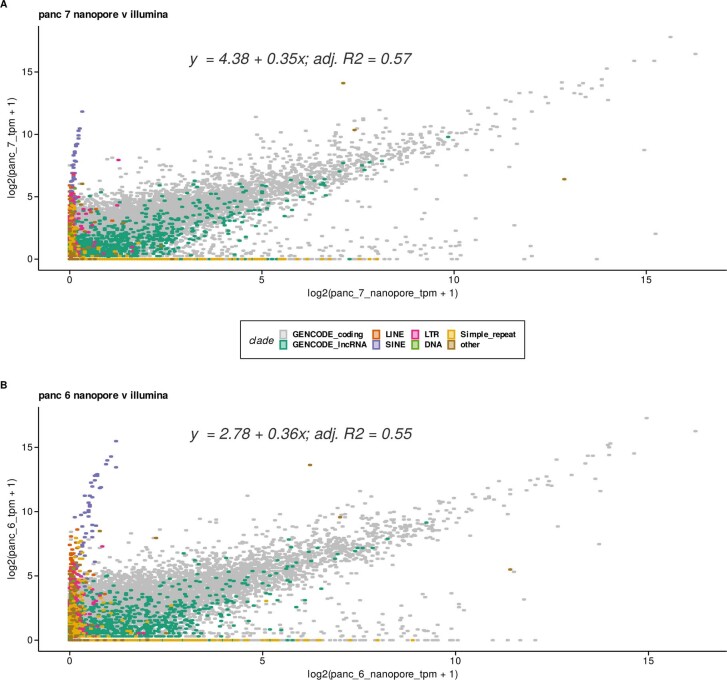
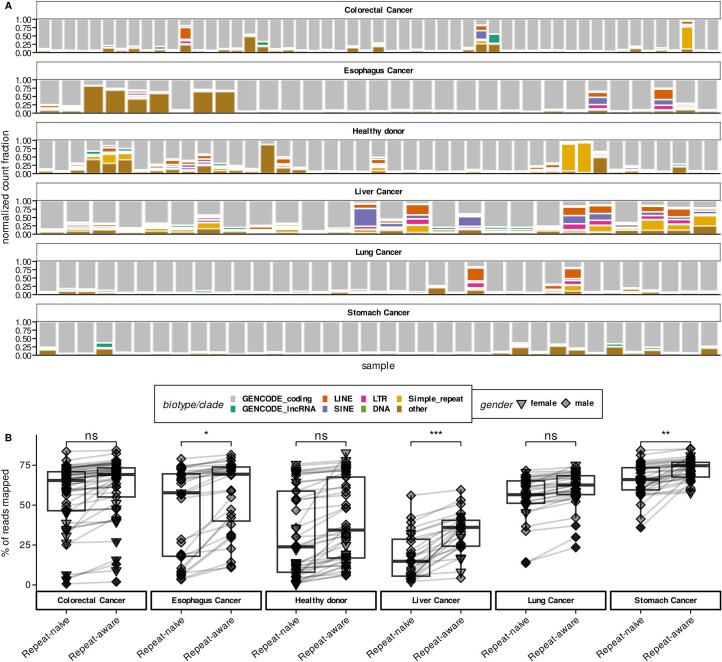
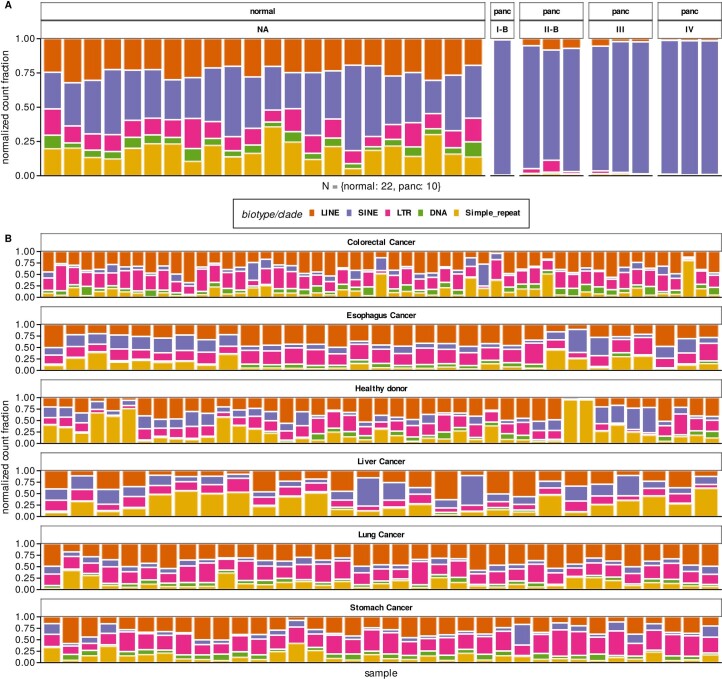
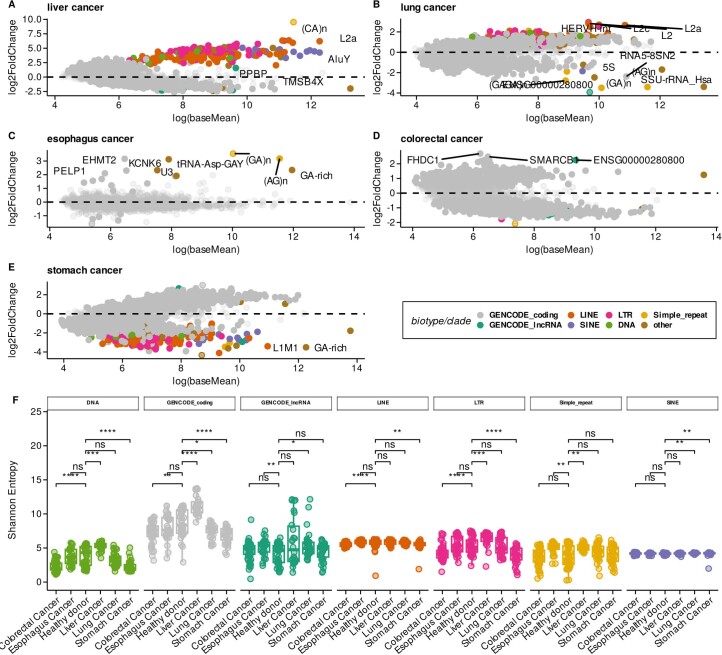
Liquid biopsies provide a means for the profiling of cell-free RNAs secreted by cells throughout the body. Although well-annotated coding and non-coding transcripts in blood are readily detectable and can serve as biomarkers of disease, the overall diagnostic utility of the cell-free transcriptome remains unclear. Here we show that RNAs derived from transposable elements and other repeat elements are enriched in the cell-free transcriptome of patients with cancer, and that they serve as signatures for the accurate classification of the disease. We used repeat-element-aware liquid-biopsy technology and single-molecule nanopore sequencing to profile the cell-free transcriptome in plasma from patients with cancer and to examine millions of genomic features comprising all annotated genes and repeat elements throughout the genome. By aggregating individual repeat elements to the subfamily level, we found that samples with pancreatic cancer are enriched with specific Alu subfamilies, whereas other cancers have their own characteristic cell-free RNA profile. Our findings show that repetitive RNA sequences are abundant in blood and can be used as disease-specific diagnostic biomarkers.
© 2023. The Author(s).
Conflict of interest statement
D.H.K. and R.E.R. are inventors on patent applications covering the methods and compositions to detect cancer using cell-free RNA submitted by the Regents of the University of California. D.H.K. and R.E.R. are founders and shareholders and D.H.K. is a board member of LincRNA Bio. S.Y.C. has served as a consultant to United Therapeutics and Acceleron Pharma. S.Y.C. has held research grants from Actelion, Bayer and Pfizer. S.Y.C. is a director, officer and shareholder of Synhale Therapeutics. S.Y.C. has submitted patent applications regarding metabolism in pulmonary hypertension.
Figures
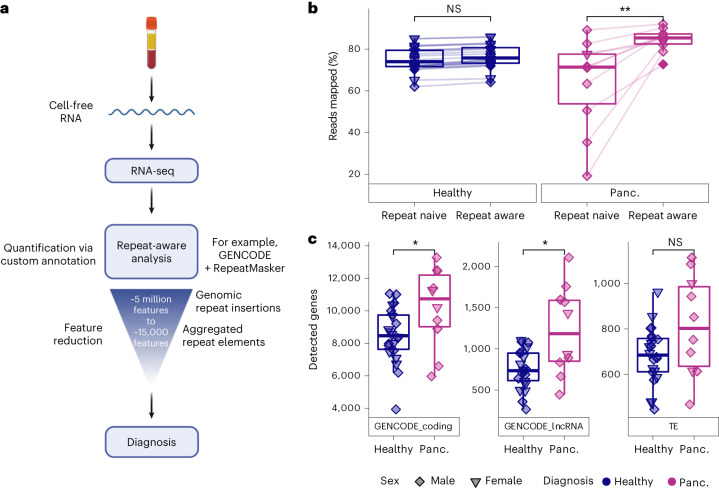
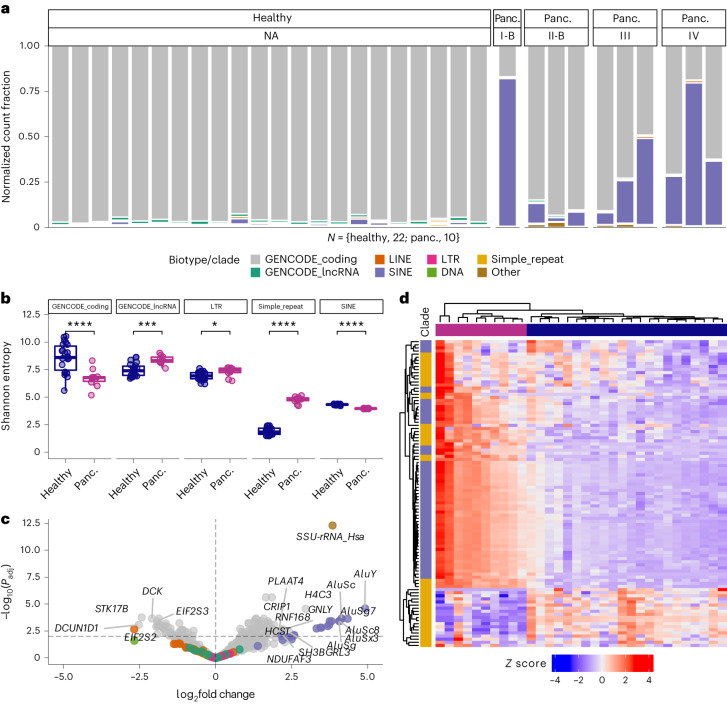
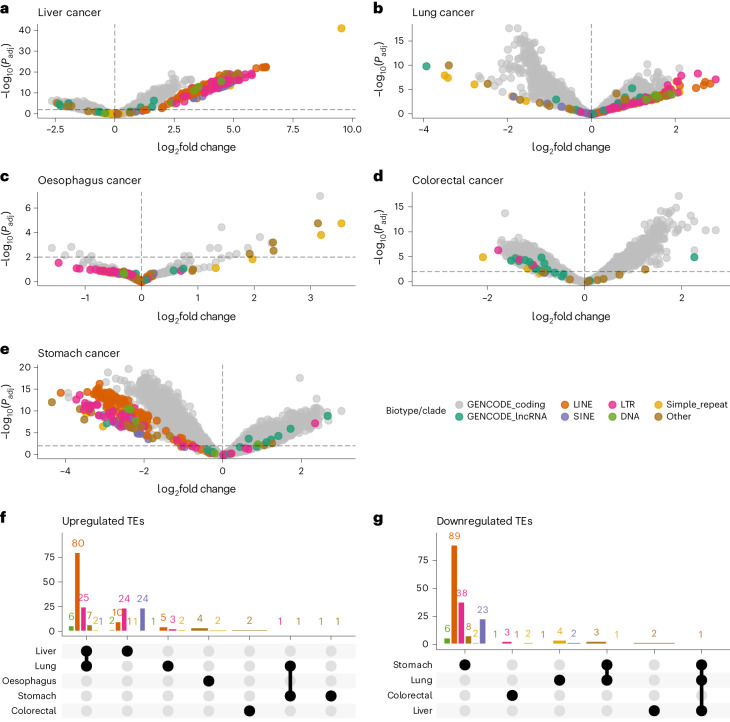
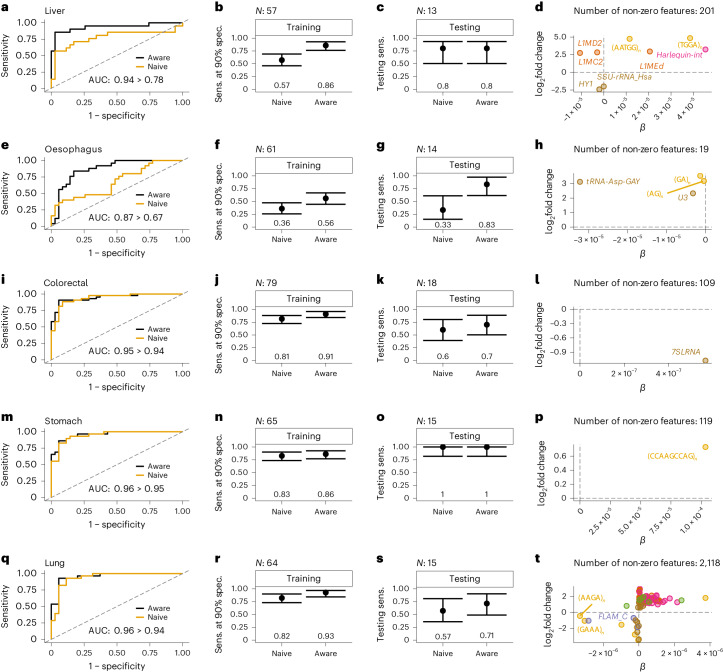
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
