

Single-cell massively-parallel multiplexed microbial sequencing (M3-seq) identifies rare bacterial populations and profiles phage infection
- PMID: 37653008
- PMCID: PMC10522482
- DOI: 10.1038/s41564-023-01462-3
Single-cell massively-parallel multiplexed microbial sequencing (M3-seq) identifies rare bacterial populations and profiles phage infection
Abstract
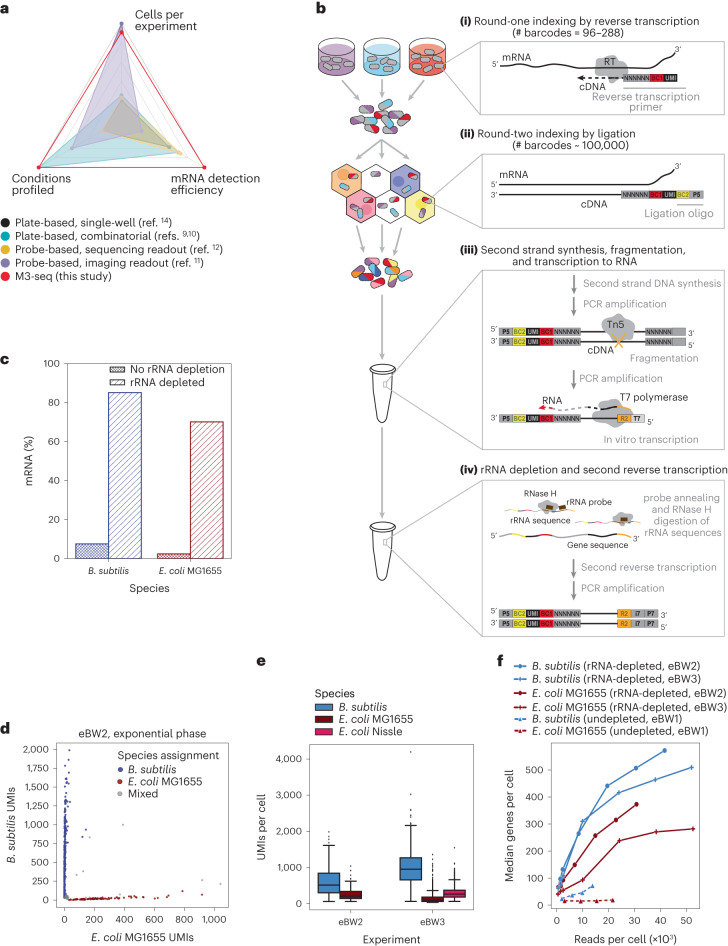
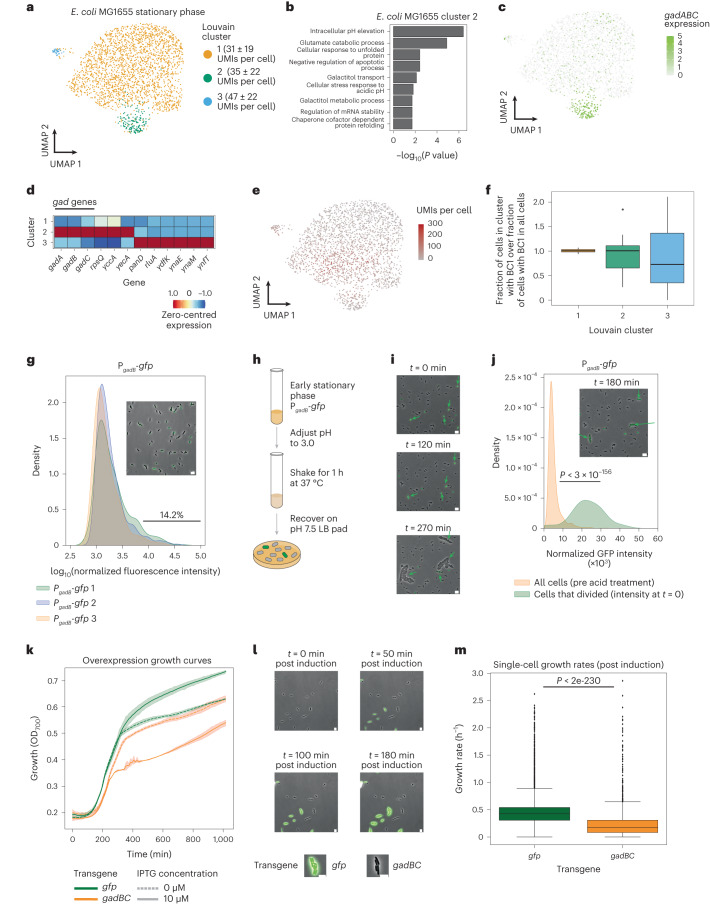
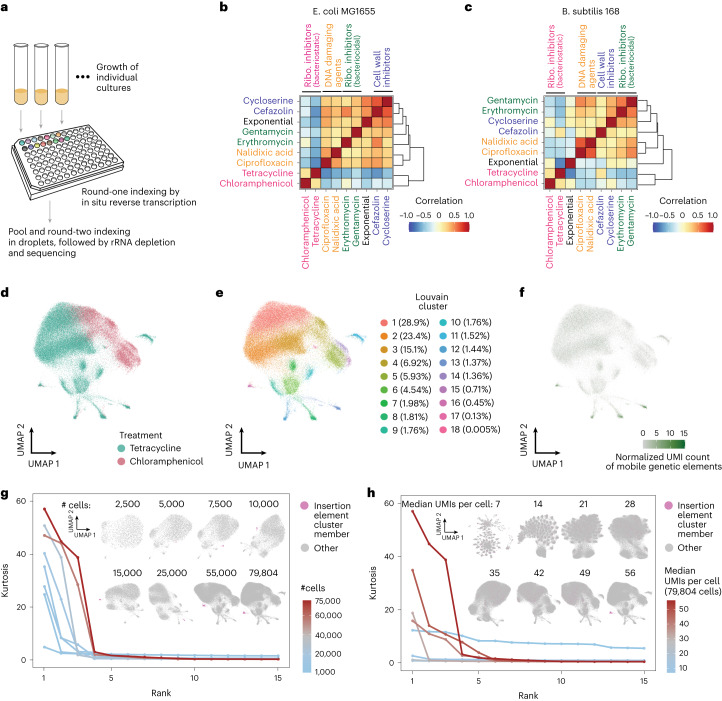
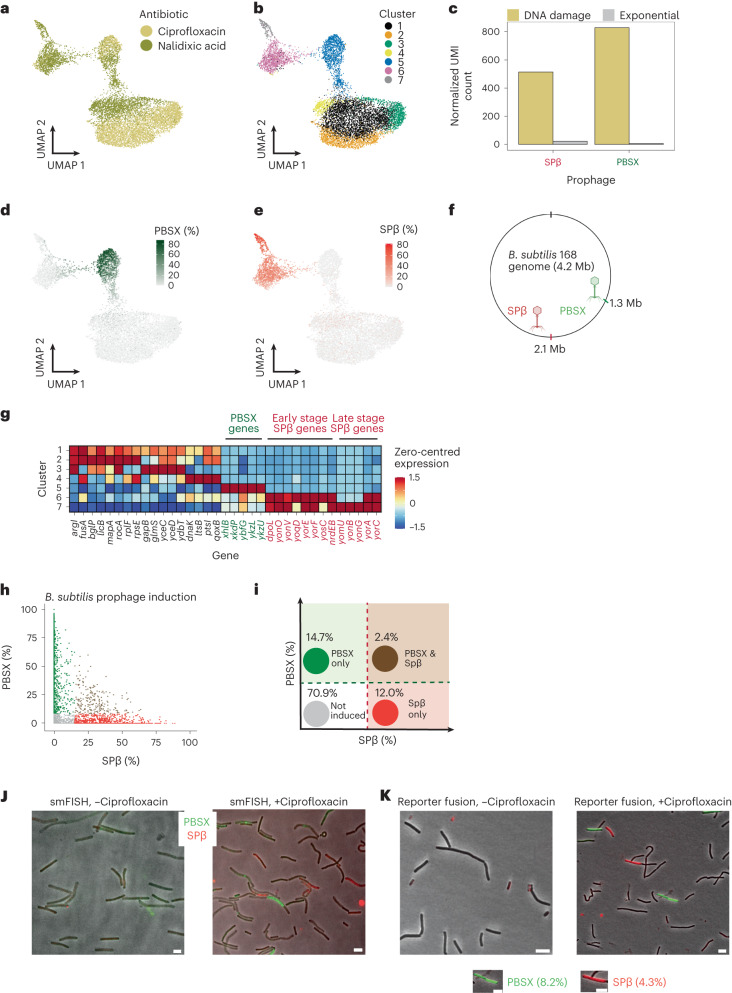
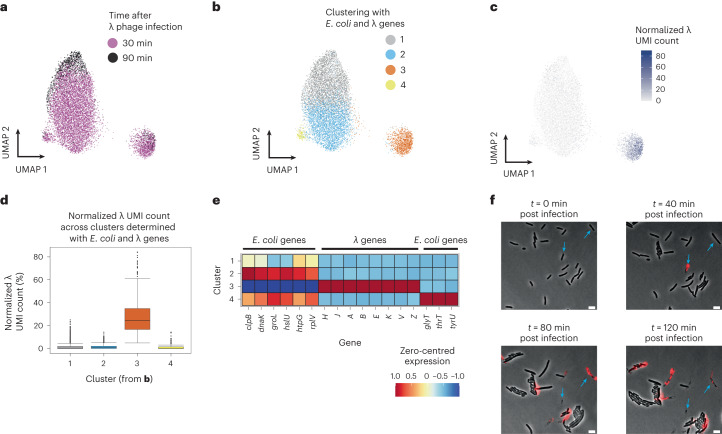
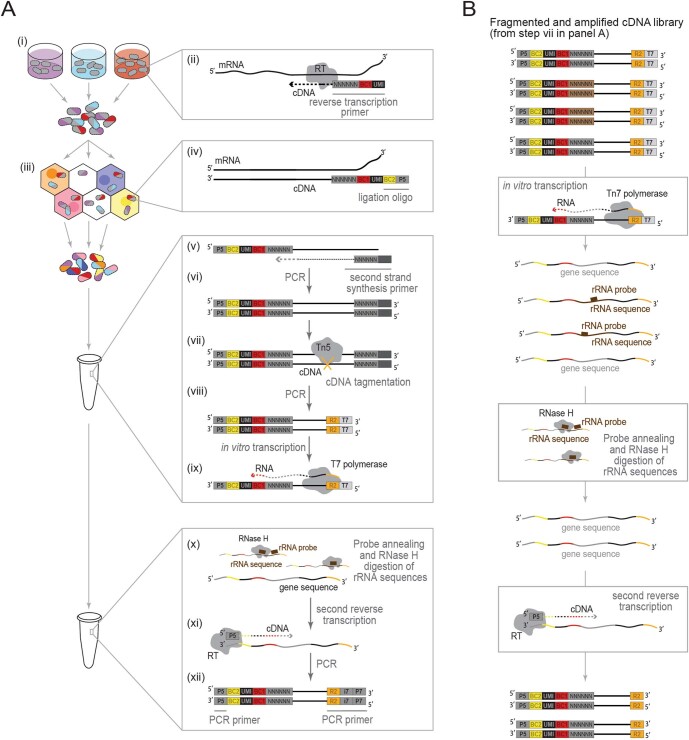
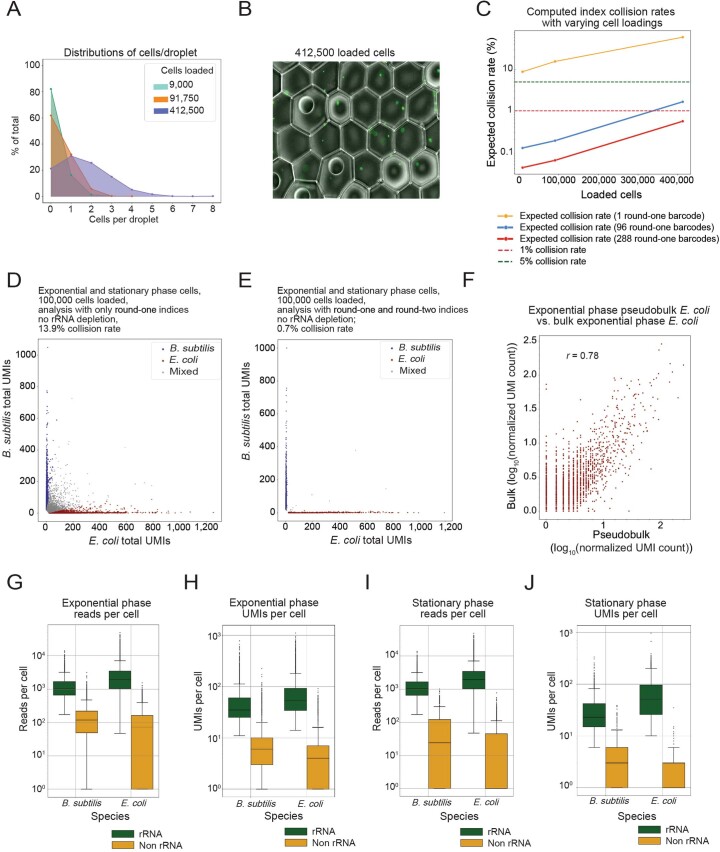
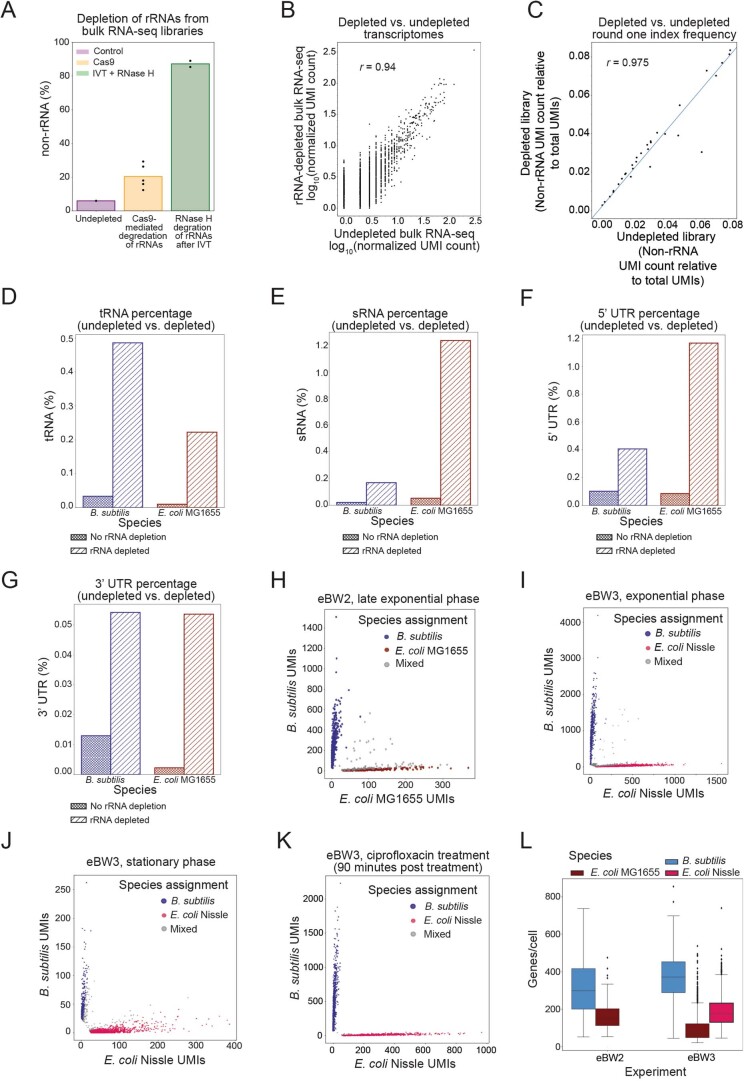
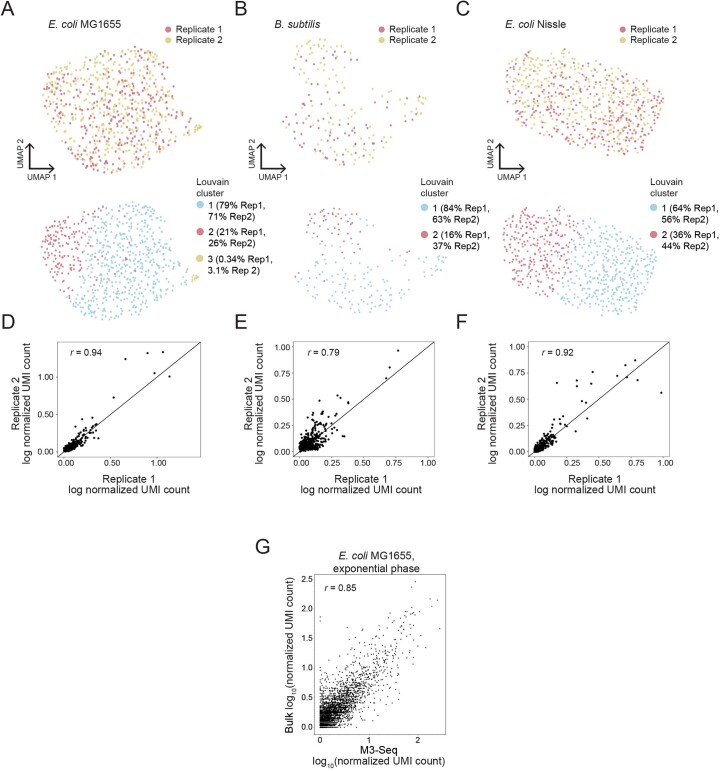
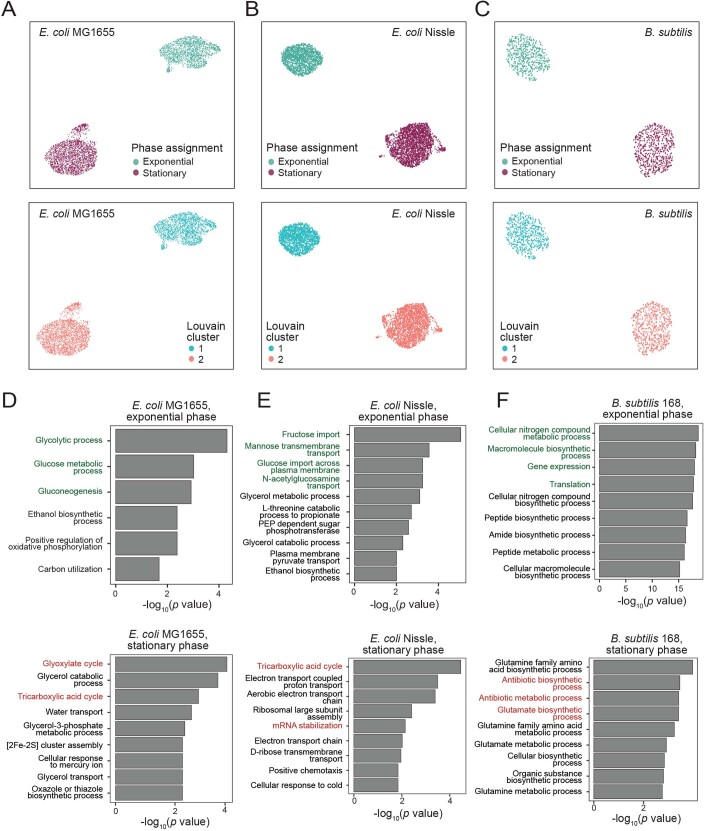
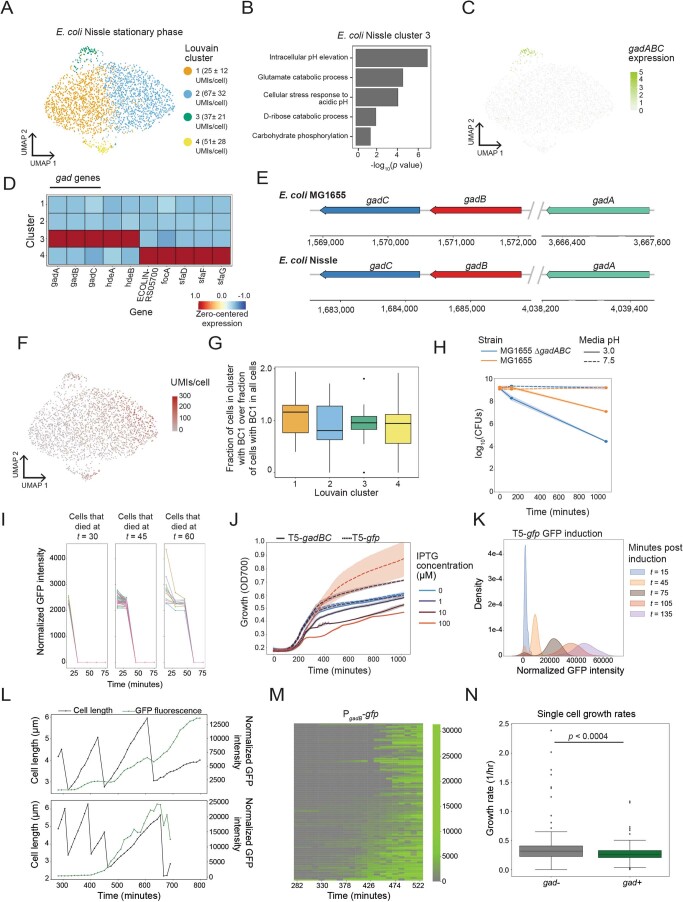
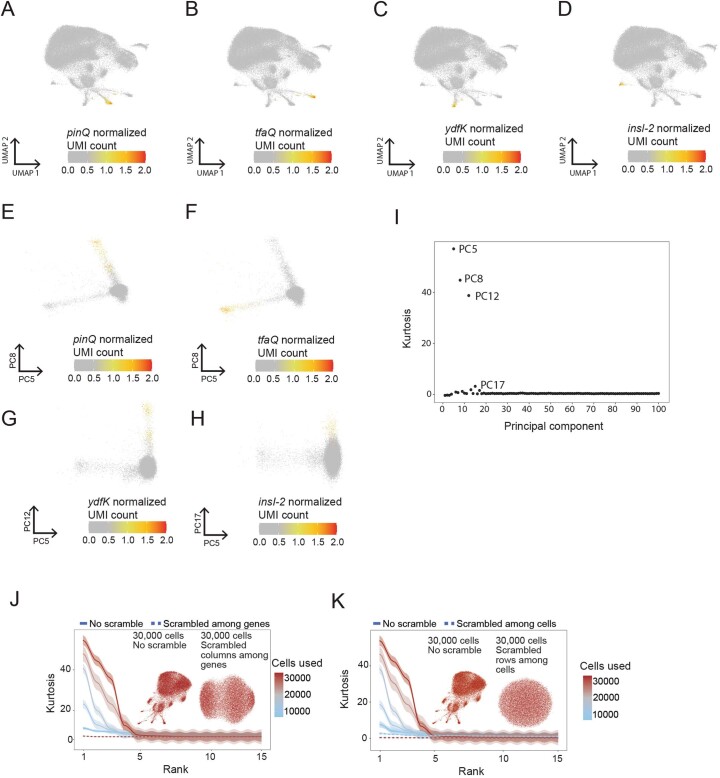
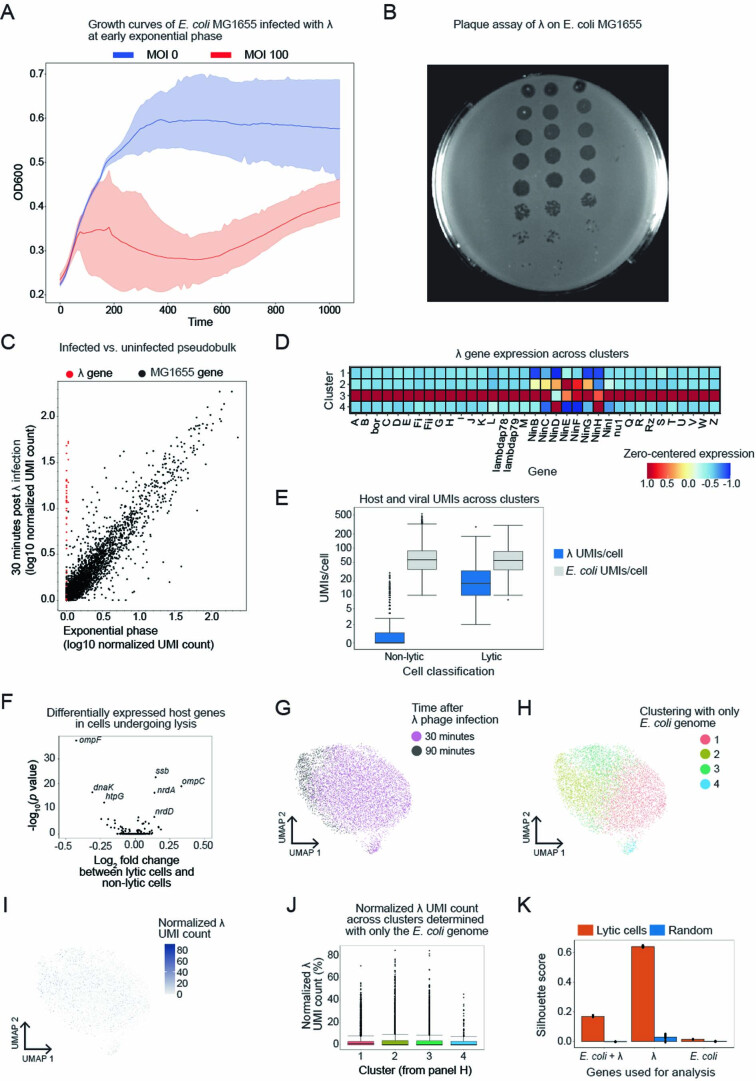
Bacterial populations are highly adaptive. They can respond to stress and survive in shifting environments. How the behaviours of individual bacteria vary during stress, however, is poorly understood. To identify and characterize rare bacterial subpopulations, technologies for single-cell transcriptional profiling have been developed. Existing approaches show some degree of limitation, for example, in terms of number of cells or transcripts that can be profiled. Due in part to these limitations, few conditions have been studied with these tools. Here we develop massively-parallel, multiplexed, microbial sequencing (M3-seq)-a single-cell RNA-sequencing platform for bacteria that pairs combinatorial cell indexing with post hoc rRNA depletion. We show that M3-seq can profile bacterial cells from different species under a range of conditions in single experiments. We then apply M3-seq to hundreds of thousands of cells, revealing rare populations and insights into bet-hedging associated with stress responses and characterizing phage infection.
© 2023. The Author(s).
Conflict of interest statement
B.A. is an advisory board member, with options, for Arbor Biotechnologies and Tessera Therapeutics and holds equity in Celsius Therapeutics. Z.G. is the founder of ArrePath. The remaining authors declare no competing interests.
Figures

Similar articles
-
Enhancing the sensitivity of bacterial single-cell RNA sequencing using RamDA-seq and Cas9-based rRNA depletion.J Biosci Bioeng. 2023 Aug;136(2):152-158. doi: 10.1016/j.jbiosc.2023.05.010. Epub 2023 Jun 11. J Biosci Bioeng. 2023. PMID: 37311684
-
High-throughput single-microbe RNA sequencing reveals adaptive state heterogeneity and host-phage activity associations in human gut microbiome.Protein Cell. 2025 Mar 8;16(3):211-226. doi: 10.1093/procel/pwae027. Protein Cell. 2025. PMID: 38779805 Free PMC article.
-
Droplet-based high-throughput single microbe RNA sequencing by smRandom-seq.Nat Commun. 2023 Aug 23;14(1):5130. doi: 10.1038/s41467-023-40137-9. Nat Commun. 2023. PMID: 37612289 Free PMC article.
-
Going viral: next-generation sequencing applied to phage populations in the human gut.Nat Rev Microbiol. 2012 Sep;10(9):607-17. doi: 10.1038/nrmicro2853. Epub 2012 Aug 6. Nat Rev Microbiol. 2012. PMID: 22864264 Free PMC article. Review.
-
Advances and challenges in single-cell RNA-seq of microbial communities.Curr Opin Microbiol. 2020 Oct;57:102-110. doi: 10.1016/j.mib.2020.10.001. Epub 2020 Nov 4. Curr Opin Microbiol. 2020. PMID: 33160164 Review.
Cited by
-
Transcriptomic profiling of individual bacteria by MATQ-seq.Nat Protoc. 2025 Apr 9. doi: 10.1038/s41596-025-01157-5. Online ahead of print. Nat Protoc. 2025. PMID: 40204970 Review.
-
Reply to: False positives in study of memory-related gene expression.Nature. 2025 Jun;642(8066):E4-E6. doi: 10.1038/s41586-025-08989-x. Nature. 2025. PMID: 40467742 No abstract available.
-
Genomic insights into bacteriophages: a new frontier in AMR detection and phage therapy.Brief Funct Genomics. 2025 Jan 15;24:elaf011. doi: 10.1093/bfgp/elaf011. Brief Funct Genomics. 2025. PMID: 40720171 Free PMC article. Review.
-
Exploitation of microbial activities at low pH to enhance planetary health.FEMS Microbiol Rev. 2024 Jan 12;48(1):fuad062. doi: 10.1093/femsre/fuad062. FEMS Microbiol Rev. 2024. PMID: 37985709 Free PMC article.
-
CRISPRi functional genomics in bacteria and its application to medical and industrial research.Microbiol Mol Biol Rev. 2024 Jun 27;88(2):e0017022. doi: 10.1128/mmbr.00170-22. Epub 2024 May 29. Microbiol Mol Biol Rev. 2024. PMID: 38809084 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
