

The stress-responsive protein REDD1 and its pathophysiological functions
- PMID: 37653030
- PMCID: PMC10545776
- DOI: 10.1038/s12276-023-01056-3
The stress-responsive protein REDD1 and its pathophysiological functions
Abstract
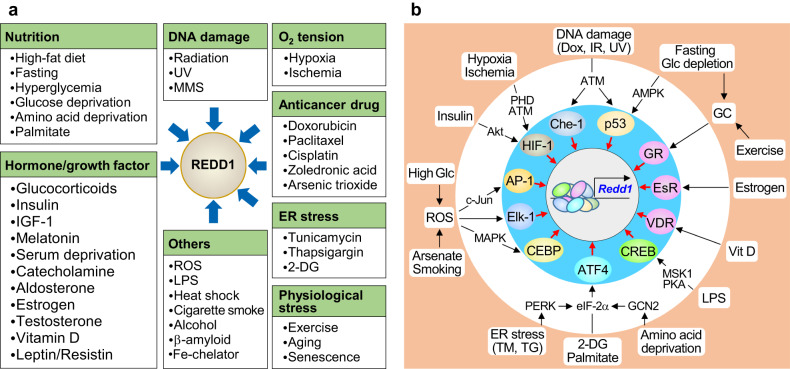
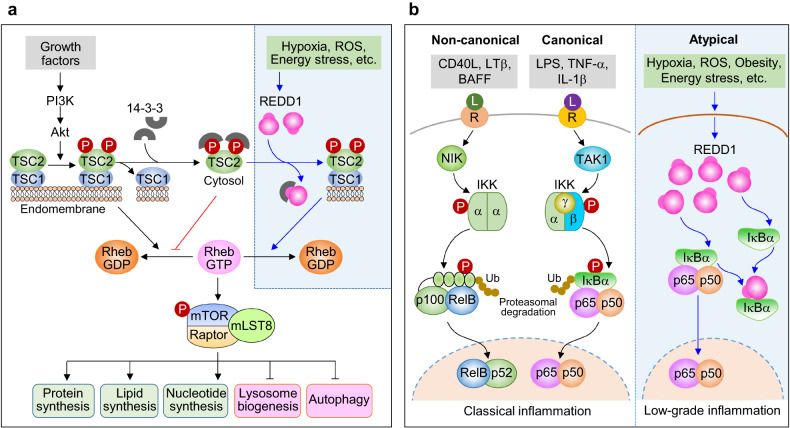
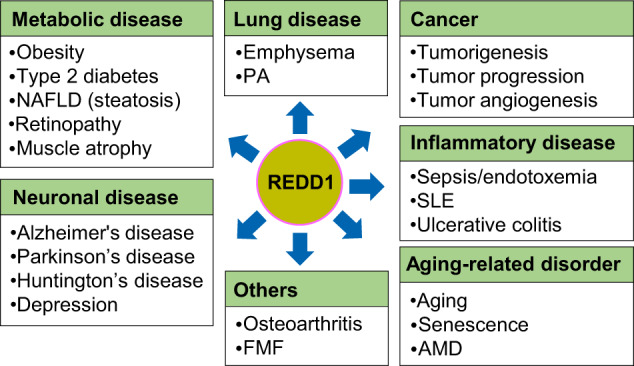
Regulated in development and DNA damage-response 1 (REDD1) is a stress-induced protein that controls various cellular functions, including metabolism, oxidative stress, autophagy, and cell fate, and contributes to the pathogenesis of metabolic and inflammatory disorders, neurodegeneration, and cancer. REDD1 usually exerts deleterious effects, including tumorigenesis, metabolic inflammation, neurodegeneration, and muscle dystrophy; however, it also exhibits protective functions by regulating multiple intrinsic cell activities through either an mTORC1-dependent or -independent mechanism. REDD1 typically regulates mTORC1 signaling, NF-κB activation, and cellular pro-oxidant or antioxidant activity by interacting with 14-3-3 proteins, IκBα, and thioredoxin-interacting protein or 75 kDa glucose-regulated protein, respectively. The diverse functions of REDD1 depend on cell type, cellular context, interaction partners, and cellular localization (e.g., mitochondria, endomembrane, or cytosol). Therefore, comprehensively understanding the molecular mechanisms and biological roles of REDD1 under pathophysiological conditions is of utmost importance. In this review, based on the published literature, we highlight and discuss the molecular mechanisms underlying the REDD1 expression and its actions, biological functions, and pathophysiological roles.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Alvarez-Garcia O, et al. Regulated in development and DNA damage response 1 deficiency impairs autophagy and mitochondrial biogenesis in articular cartilage and increases the severity of experimental osteoarthritis. Arthritis Rheumatol. 2017;69:1418–1428. doi: 10.1002/art.40104. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
