

Autotaxin suppresses cytotoxic T cells via LPAR5 to promote anti-PD-1 resistance in non-small cell lung cancer
- PMID: 37655662
- PMCID: PMC10471170
- DOI: 10.1172/JCI163128
Autotaxin suppresses cytotoxic T cells via LPAR5 to promote anti-PD-1 resistance in non-small cell lung cancer
Abstract
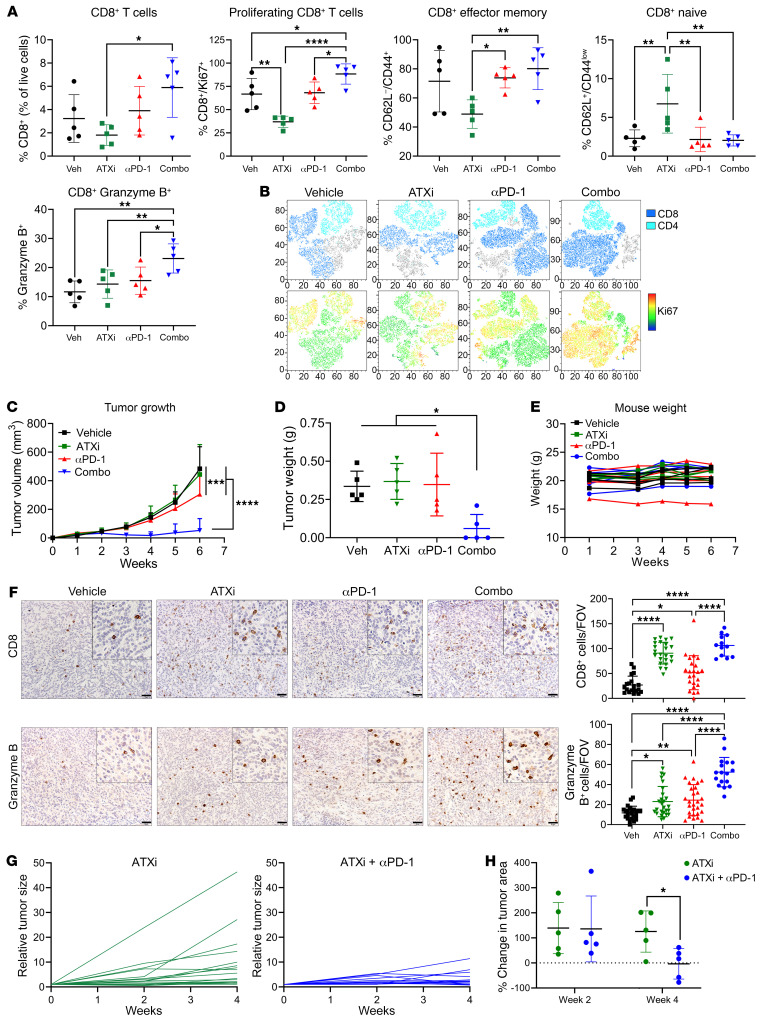
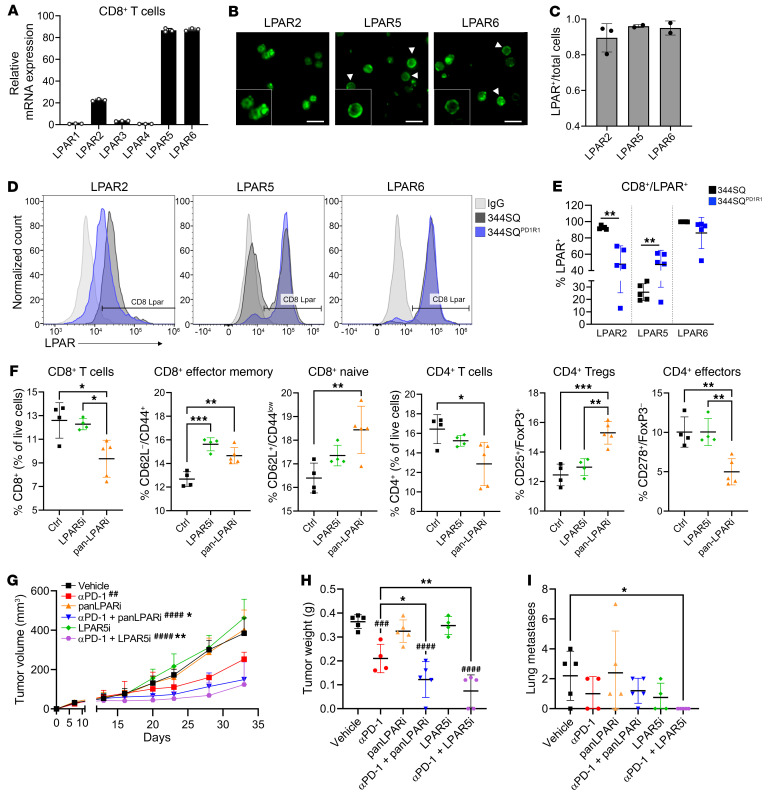
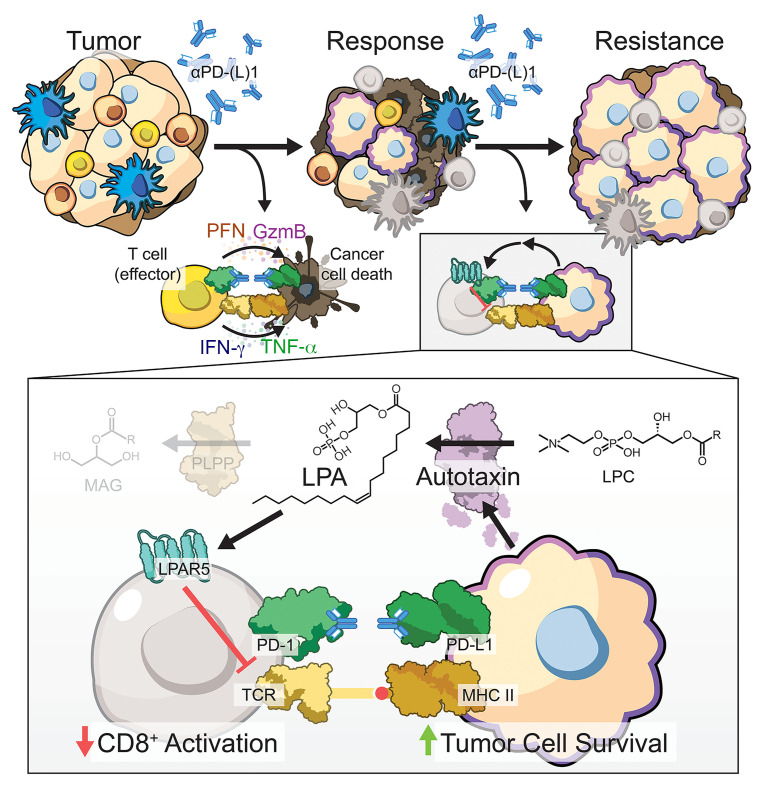
Non-small cell lung cancers that harbor concurrent KRAS and TP53 (KP) mutations are immunologically warm tumors with partial responsiveness to anti-PD-(L)1 blockade; however, most patients observe little or no durable clinical benefit. To identify novel tumor-driven resistance mechanisms, we developed a panel of KP murine lung cancer models with intrinsic resistance to anti-PD-1 and queried differential gene expression between these tumors and anti-PD-1-sensitive tumors. We found that the enzyme autotaxin (ATX), and the metabolite it produces, lysophosphatidic acid (LPA), were significantly upregulated in resistant tumors and that ATX directly modulated antitumor immunity, with its expression negatively correlating with total and effector tumor-infiltrating CD8+ T cells. Pharmacological inhibition of ATX, or the downstream receptor LPAR5, in combination with anti-PD-1 was sufficient to restore the antitumor immune response and efficaciously control lung tumor growth in multiple KP tumor models. Additionally, ATX was significantly correlated with inflammatory gene signatures, including a CD8+ cytolytic score in multiple lung adenocarcinoma patient data sets, suggesting that an activated tumor-immune microenvironment upregulates ATX and thus provides an opportunity for cotargeting to prevent acquired resistance to anti-PD-1 treatment. These data reveal the ATX/LPA axis as an immunosuppressive pathway that diminishes the immune checkpoint blockade response in lung cancer.
Keywords: Cancer immunotherapy; Immunology; Lung cancer; Oncology; Phosphodiesterases.
Conflict of interest statement
Figures
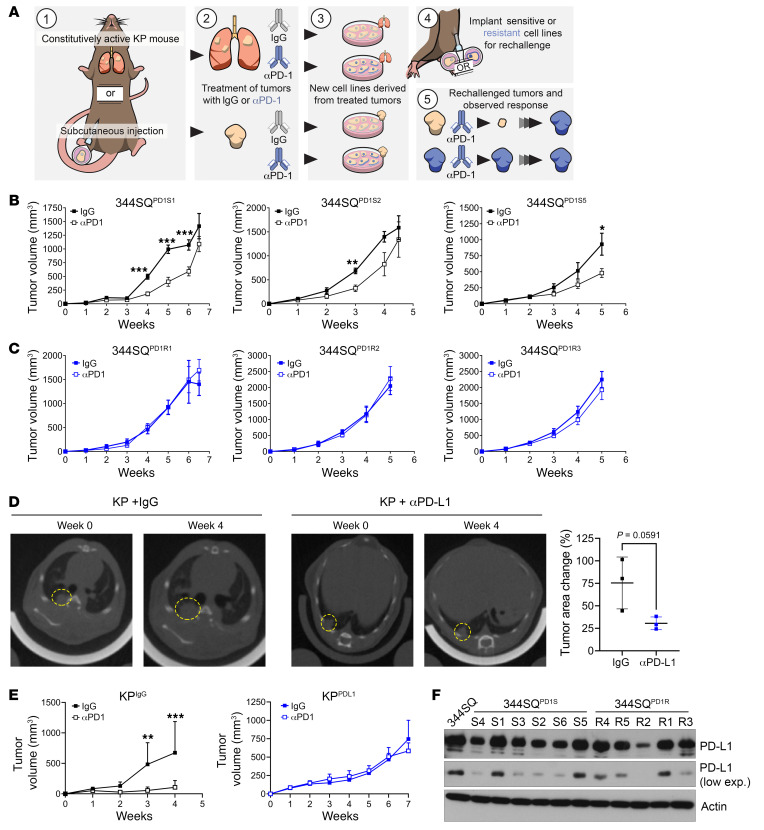
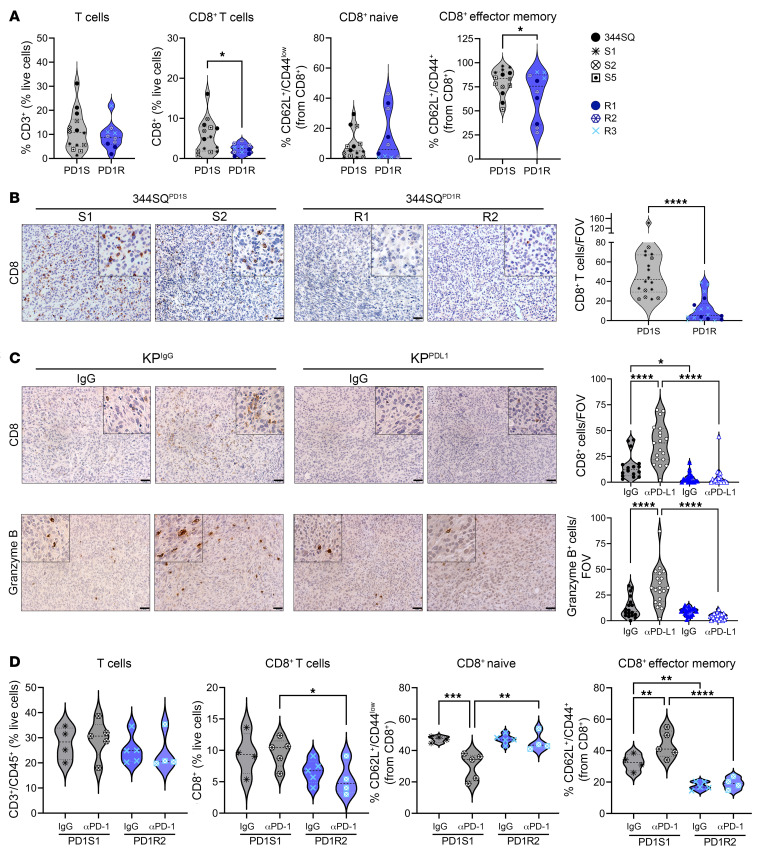
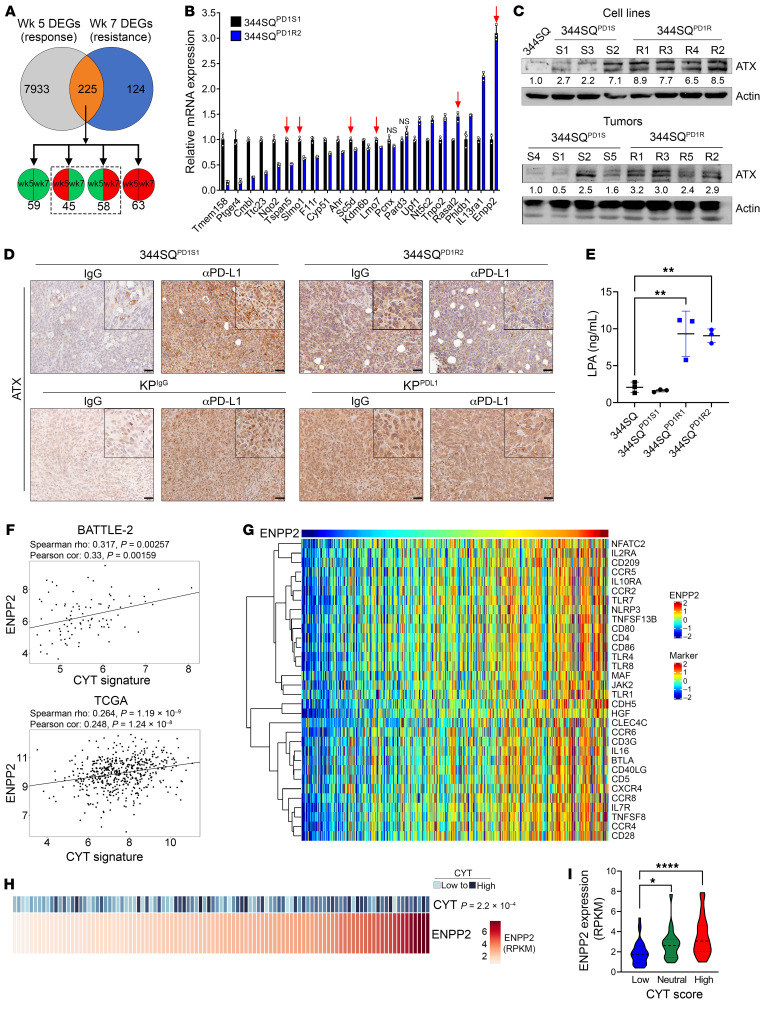
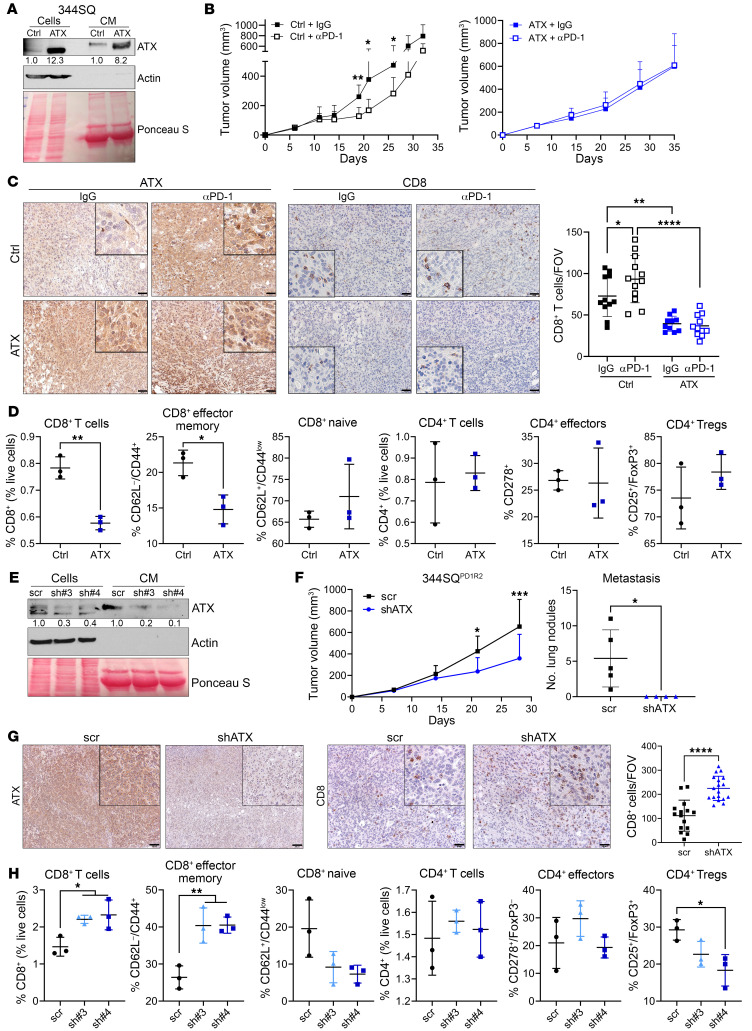
References
-
- Surveillance, Epidemiology, and End Results Program. Stat Fact Sheets: Lung and Bronchus. http://seer.cancer.gov/statfacts/html/lungb.html Accessed June 14, 2022.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
