

Fungal co-expression network analyses identify pathogen gene modules associated with host insect invasion
- PMID: 37656157
- PMCID: PMC10581046
- DOI: 10.1128/spectrum.01809-23
Fungal co-expression network analyses identify pathogen gene modules associated with host insect invasion
Abstract
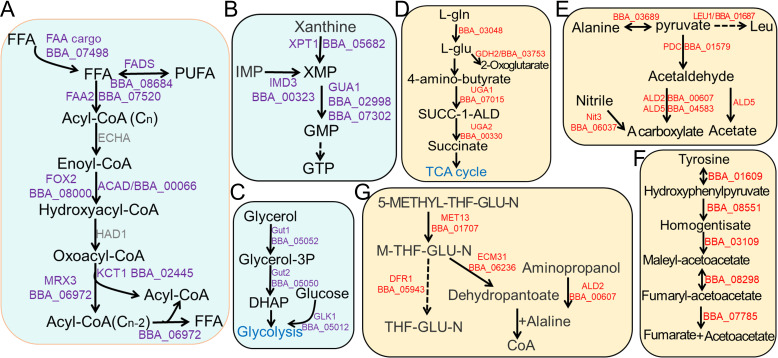
The broad host range fungal insect pathogen, Beauveria bassiana, has been commercialized as an alternative to chemical insecticides for pest control worldwide. B. bassiana represents a unique model system with which to examine host-pathogen interactions, and a wide range of genes and processes have been studied. However, significant aspects of virulence, particularly on the genomic scale, remain poorly studied. Here, we have combined available transcriptomes with three newly generated data sets for a combined total analysis of 76 deep-sequenced samples covering growth, development, stress responses, and infection during the life cycle of B. bassiana. Co-expression network analyses resulted in the identification of gene modules enriched during two critical stages of the infection process, namely (i) cuticle penetration and (ii) in vivo hyphal body (dimorphic transition) growth capable of avoiding innate and humoral immune defenses. These analyses identify unique signatures of metabolism, signaling, secondary metabolite production, host defense suppression, membrane reorganization, effector production, and secretion for each stage, including genetic regulators and epigenetic patterns. These data provide a comprehensive framework for understanding and probing fungal adaptations to its pathogenic life cycle and expand the candidate repertoire for continued dissection of the host-pathogen interaction. IMPORTANCE Insect fungal pathogens have evolved unique strategies for overcoming host structural and immunological defenses that span from the sclerotized cuticle to innate and humoral cellular responses. Two critical stages of the infection process involve (i) cuticle penetration and (ii) immune evasion within the insect hemocoel. A set of 76 global transcriptomic data for B. bassiana that include the cuticle penetration and hemocoel growth stages were analyzed for patterns (gene modules) of expression, yielding unique insights into these different life stages. These analyses integrate gene networks involved in fungal development, stress response and pathogenesis to further the systematic understanding of the global processes integral to the unique adaptation employed by fungal pathogens of insects.
Keywords: Beauveria bassiana; co-expression; entomopathogenic fungi; host-pathogen interaction; infection-associated modules; transcriptome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures









References
-
- Skinner M, Parker BL, Kim JS. 2014. Chapter 10 - Role of entomopathogenic fungi in integrated pest management, p 169–191. In Abrol DP (ed), J Integr pest Manag. Academic Press, San Diego.
-
- Chandler D. 2017. Chapter 5 - Basic and applied research on entomopathogenic fungi, p 69-89. In Lacey LA (ed), Microbial control of insect and mite pests. Academic Press.
-
- Peng G, Xie J, Guo R, Keyhani NO, Zeng D, Yang P, Xia Y. 2021. Long-term field evaluation and large-scale application of a Metarhizium anisopliae strain for controlling major rice pests. J Pest Sci 94:969–980. doi: 10.1007/s10340-020-01313-8 - DOI
-
- Wang CS, Feng MG. 2014. Advances in fundamental and applied studies in China of fungal biocontrol agents for use against arthropod pests. Biol Control 68:129–135. doi: 10.1016/j.biocontrol.2013.06.017 - DOI
LinkOut - more resources
Full Text Sources
