

Perspectives on Ligand/Protein Binding Kinetics Simulations: Force Fields, Machine Learning, Sampling, and User-Friendliness
- PMID: 37656199
- PMCID: PMC10536999
- DOI: 10.1021/acs.jctc.3c00641
Perspectives on Ligand/Protein Binding Kinetics Simulations: Force Fields, Machine Learning, Sampling, and User-Friendliness
Abstract
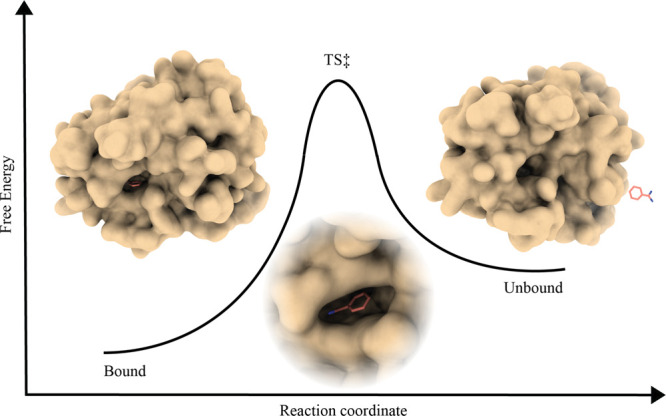
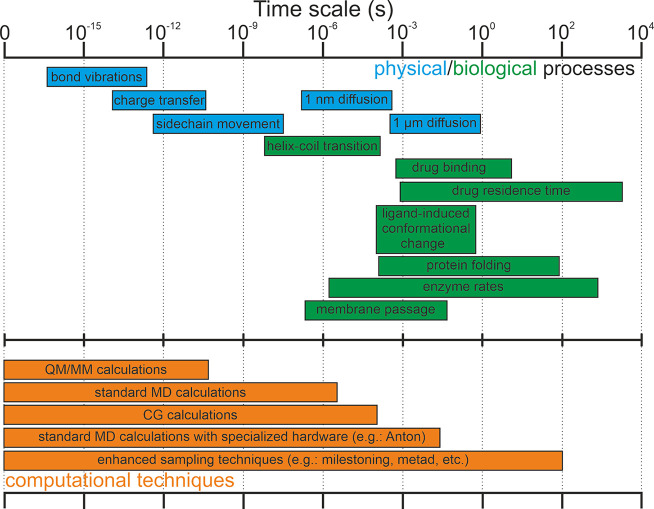
Computational techniques applied to drug discovery have gained considerable popularity for their ability to filter potentially active drugs from inactive ones, reducing the time scale and costs of preclinical investigations. The main focus of these studies has historically been the search for compounds endowed with high affinity for a specific molecular target to ensure the formation of stable and long-lasting complexes. Recent evidence has also correlated the in vivo drug efficacy with its binding kinetics, thus opening new fascinating scenarios for ligand/protein binding kinetic simulations in drug discovery. The present article examines the state of the art in the field, providing a brief summary of the most popular and advanced ligand/protein binding kinetics techniques and evaluating their current limitations and the potential solutions to reach more accurate kinetic models. Particular emphasis is put on the need for a paradigm change in the present methodologies toward ligand and protein parametrization, the force field problem, characterization of the transition states, the sampling issue, and algorithms' performance, user-friendliness, and data openness.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

References
-
- Mullard A. New drugs cost US$2.6 billion to develop. Nat. Rev. Drug Discovery 2014, 13, 877. 10.1038/nrd4507. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
