

Protocol for high-throughput semi-automated label-free- or TMT-based phosphoproteome profiling
- PMID: 37659085
- PMCID: PMC10491724
- DOI: 10.1016/j.xpro.2023.102536
Protocol for high-throughput semi-automated label-free- or TMT-based phosphoproteome profiling
Abstract
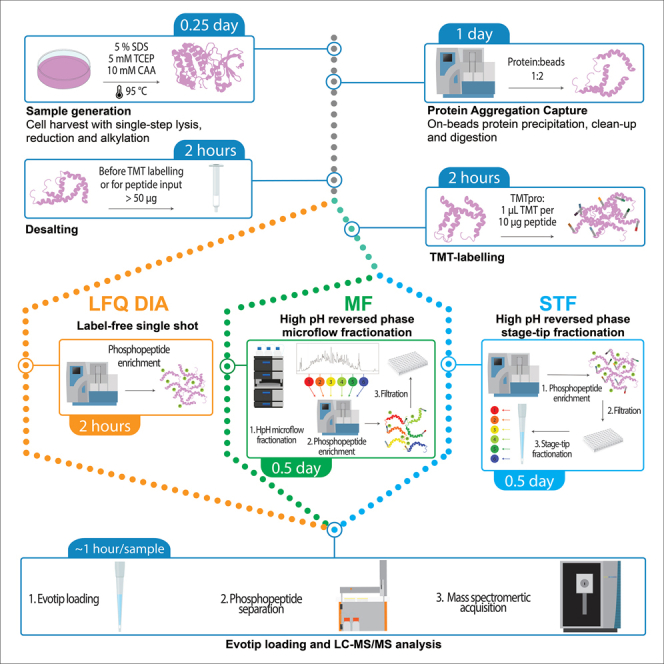
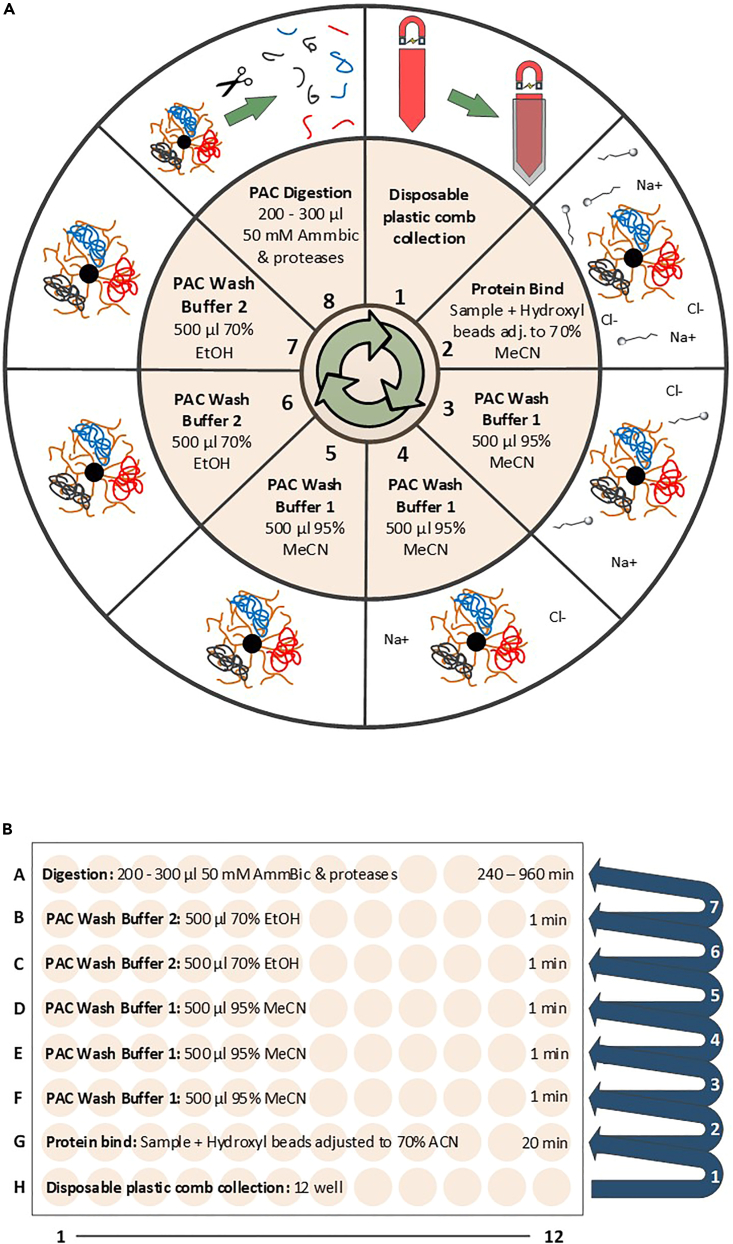
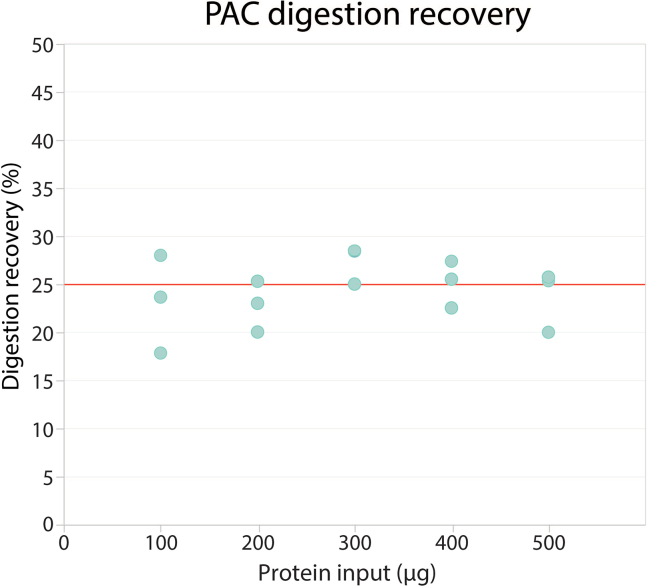
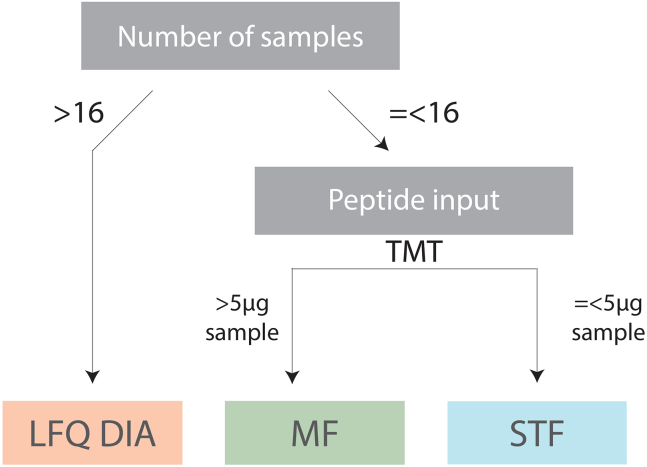
Tandem mass tags data-dependent acquisition (TMT-DDA) as well as data-independent acquisition-based label-free quantification (LFQ-DIA) have become the leading workflows to achieve deep proteome and phosphoproteome profiles. We present a modular pipeline for TMT-DDA and LFQ-DIA that integrates steps to perform scalable phosphoproteome profiling, including protein lysate extraction, clean-up, digestion, phosphopeptide enrichment, and TMT-labeling. We also detail peptide and/or phosphopeptide fractionation and pre-mass spectrometry desalting and provide researchers guidance on choosing the best workflow based on sample number and input. For complete details on the use and execution of this protocol, please refer to Koenig et al.1 and Martínez-Val et al.2.
Keywords: Mass Spectrometry; Protein Biochemistry; Proteomics.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests P.N., S.S., and J.J. work for ReSyn Biosciences that supplied some of the reagents for digest preparation and phosphopeptide enrichment utilized in this workflow. S.S. further works for Evosep Biosystems whose LC system is utilized as part of the LC-MS setup.
Figures
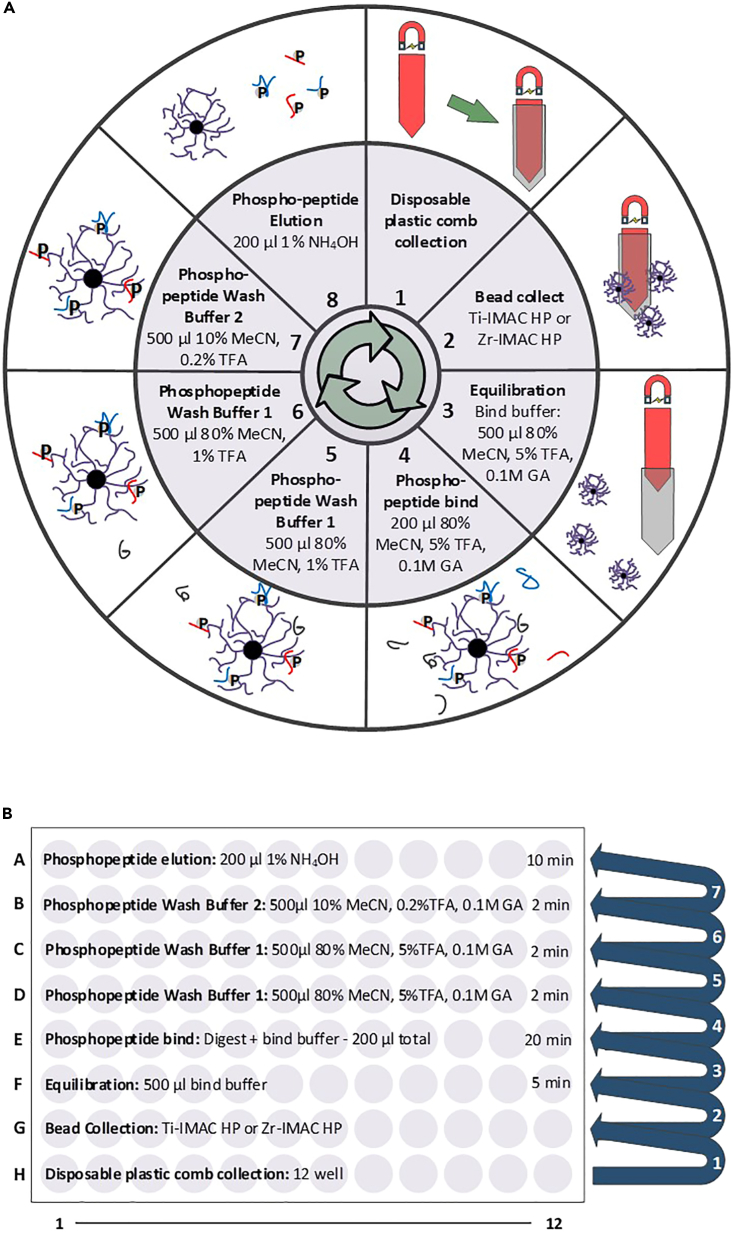
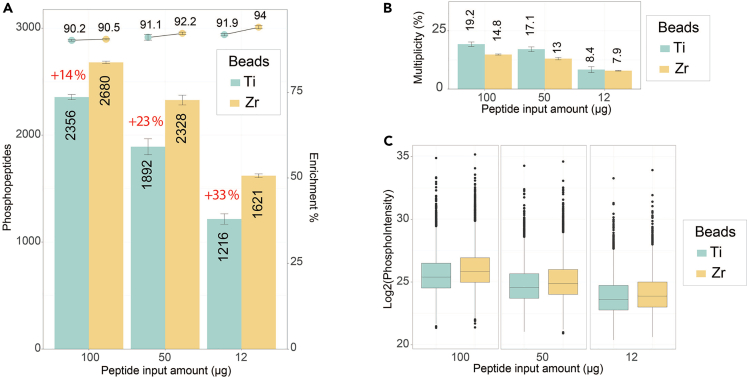
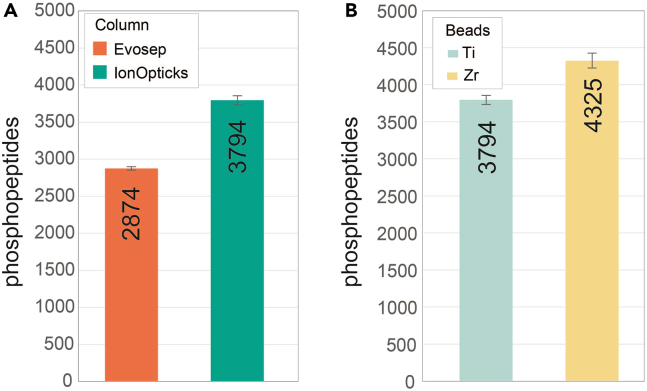
References
-
- Martínez-Val A., Fort K., Koenig C., Van der Hoeven L., Franciosa G., Moehring T., Ishihama Y., Chen Y.J., Makarov A., Xuan Y., Olsen J.V. Hybrid-DIA: intelligent data acquisition integrates targeted and discovery proteomics to analyze phospho-signaling in single spheroids. Nat. Commun. 2023;14:3599. doi: 10.1038/s41467-023-39347-y. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
