

Interpreting biologically informed neural networks for enhanced proteomic biomarker discovery and pathway analysis
- PMID: 37660105
- PMCID: PMC10475049
- DOI: 10.1038/s41467-023-41146-4
Interpreting biologically informed neural networks for enhanced proteomic biomarker discovery and pathway analysis
Abstract
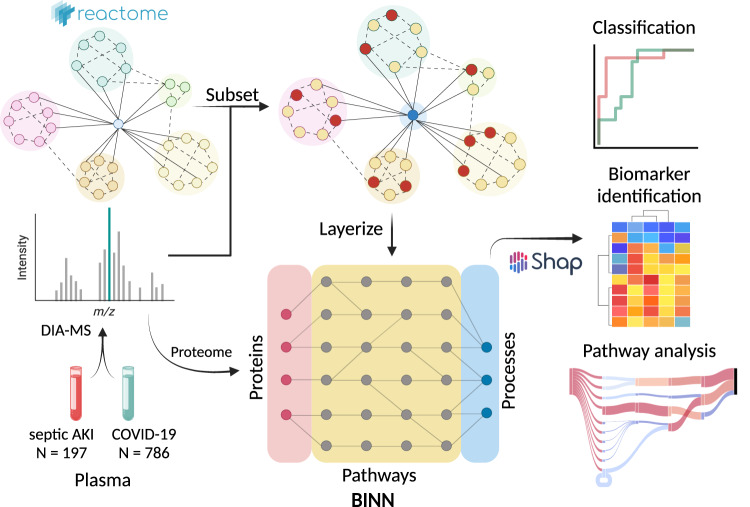
The incorporation of machine learning methods into proteomics workflows improves the identification of disease-relevant biomarkers and biological pathways. However, machine learning models, such as deep neural networks, typically suffer from lack of interpretability. Here, we present a deep learning approach to combine biological pathway analysis and biomarker identification to increase the interpretability of proteomics experiments. Our approach integrates a priori knowledge of the relationships between proteins and biological pathways and biological processes into sparse neural networks to create biologically informed neural networks. We employ these networks to differentiate between clinical subphenotypes of septic acute kidney injury and COVID-19, as well as acute respiratory distress syndrome of different aetiologies. To gain biological insight into the complex syndromes, we utilize feature attribution-methods to introspect the networks for the identification of proteins and pathways important for distinguishing between subtypes. The algorithms are implemented in a freely available open source Python-package ( https://github.com/InfectionMedicineProteomics/BINN ).
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
