

Epigenetic inheritance is unfaithful at intermediately methylated CpG sites
- PMID: 37660134
- PMCID: PMC10475082
- DOI: 10.1038/s41467-023-40845-2
Epigenetic inheritance is unfaithful at intermediately methylated CpG sites
Abstract
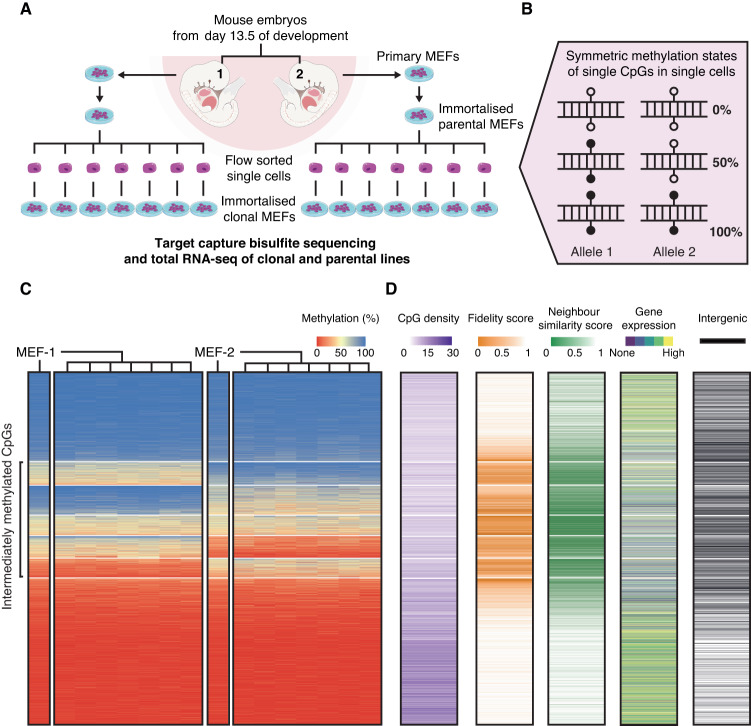
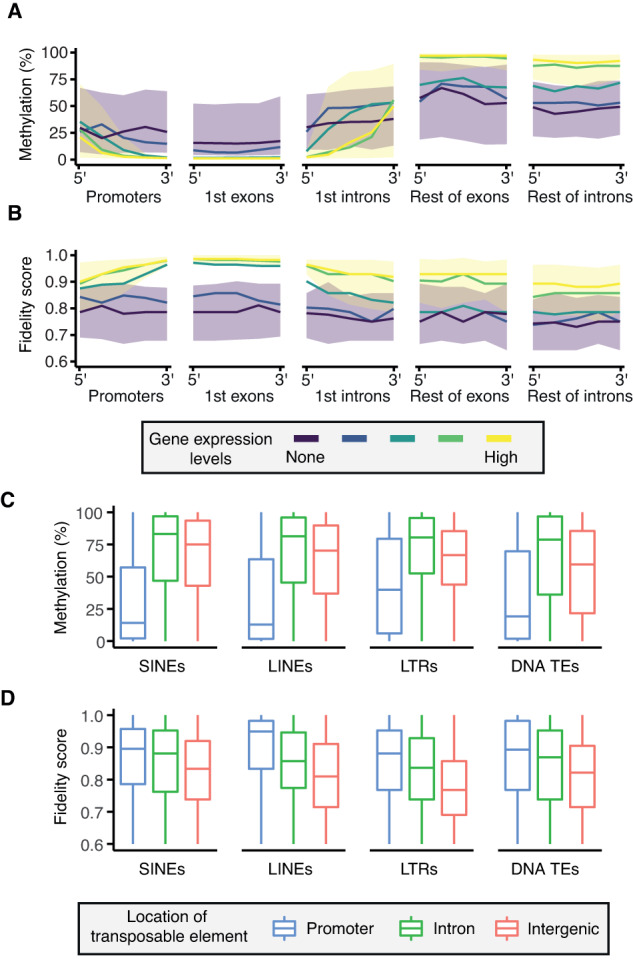
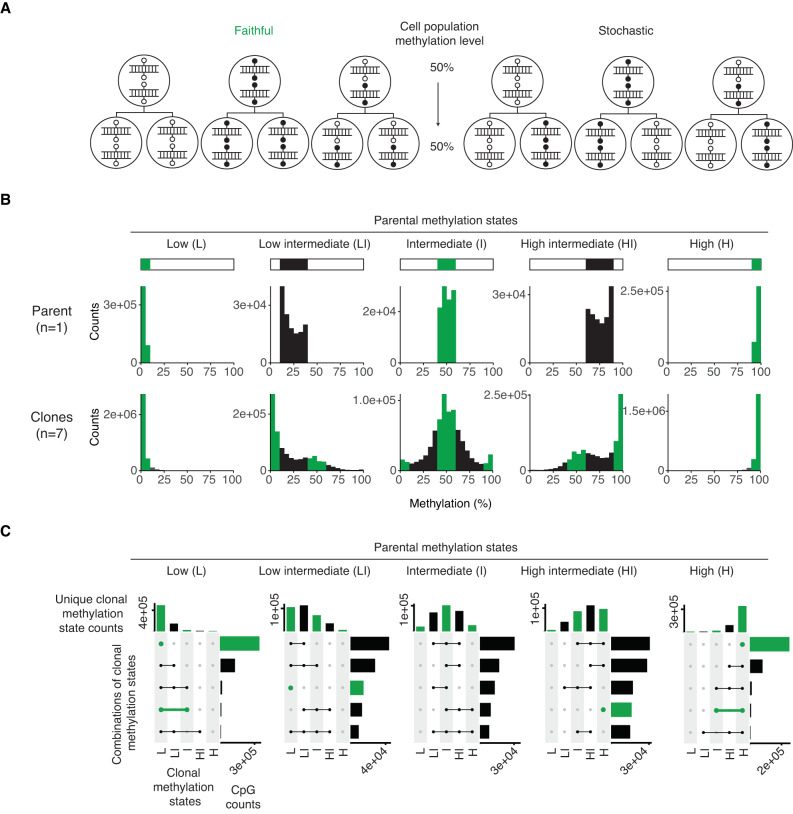
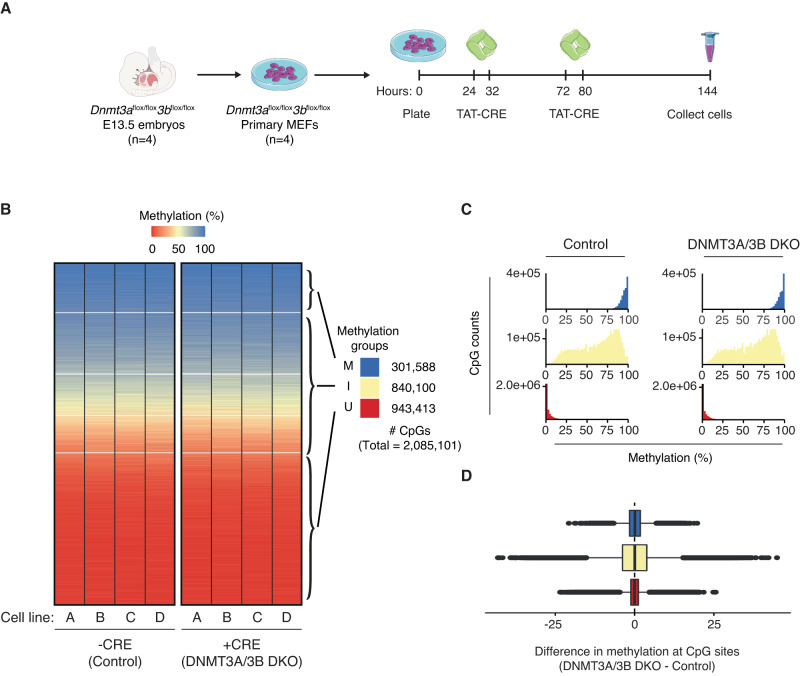
DNA methylation at the CpG dinucleotide is considered a stable epigenetic mark due to its presumed long-term inheritance through clonal expansion. Here, we perform high-throughput bisulfite sequencing on clonally derived somatic cell lines to quantitatively measure methylation inheritance at the nucleotide level. We find that although DNA methylation is generally faithfully maintained at hypo- and hypermethylated sites, this is not the case at intermediately methylated CpGs. Low fidelity intermediate methylation is interspersed throughout the genome and within genes with no or low transcriptional activity, and is not coordinately maintained between neighbouring sites. We determine that the probabilistic changes that occur at intermediately methylated sites are likely due to DNMT1 rather than DNMT3A/3B activity. The observed lack of clonal inheritance at intermediately methylated sites challenges the current epigenetic inheritance model and has direct implications for both the functional relevance and general interpretability of DNA methylation as a stable epigenetic mark.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Robertson KD. DNA methylation and human disease. Nat. Rev. Genet. 2005;6:597–610. - PubMed
-
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 2013;14:204–220. - PubMed
-
- Holliday R. DNA methylation and epigenetic inheritance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990;326:329–338. - PubMed
-
- Bestor TH, Tycko B. Creation of genomic methylation patterns. Nat. Genet. 1996;12:363–367. - PubMed
-
- Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009;10:192–206. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
