

Symmetric inheritance of parental histones governs epigenome maintenance and embryonic stem cell identity
- PMID: 37666988
- PMCID: PMC10484787
- DOI: 10.1038/s41588-023-01476-x
Symmetric inheritance of parental histones governs epigenome maintenance and embryonic stem cell identity
Abstract
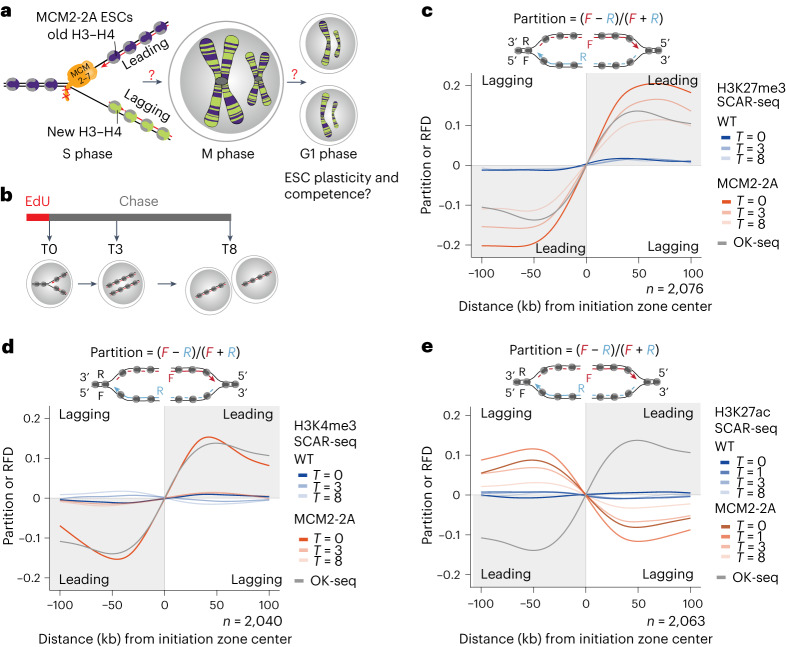
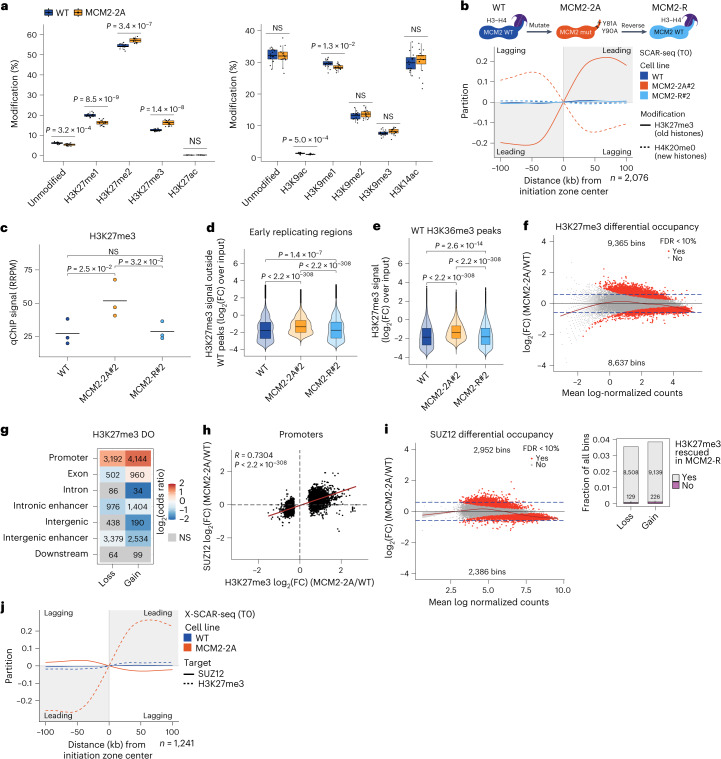
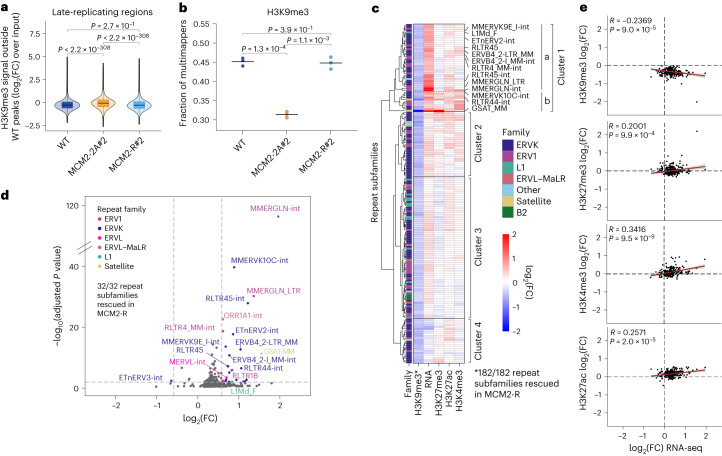
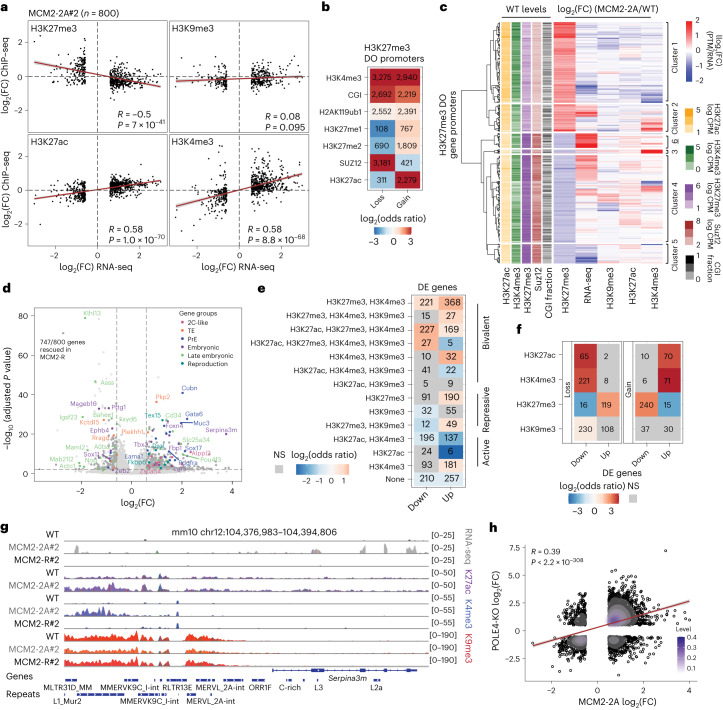
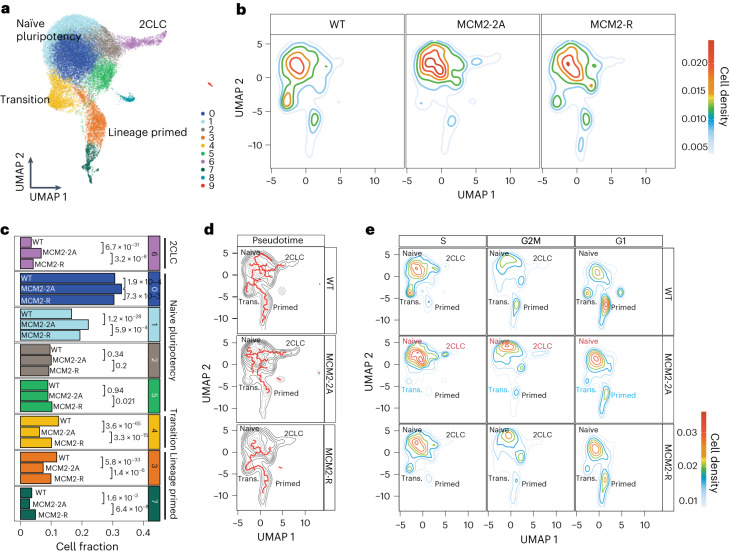
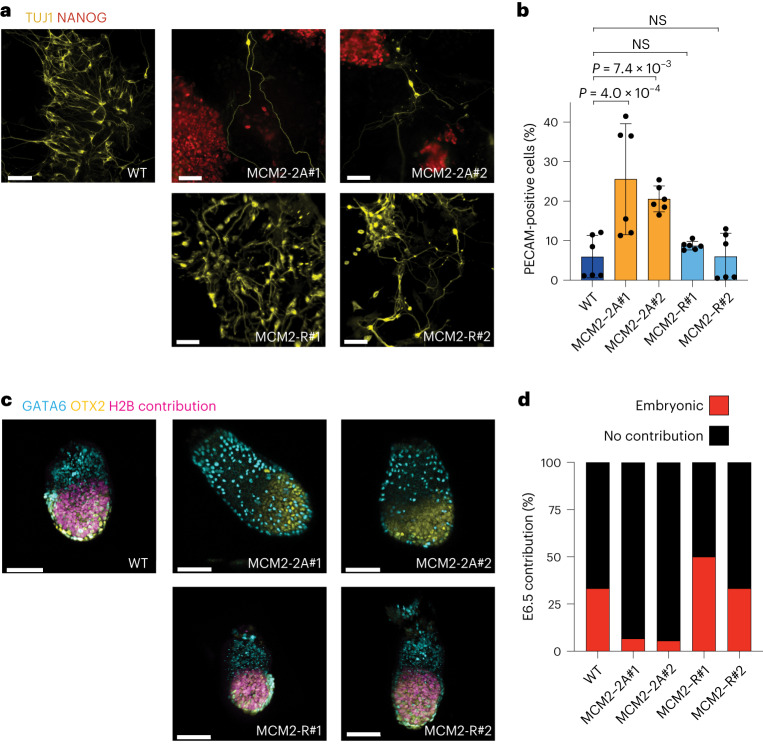
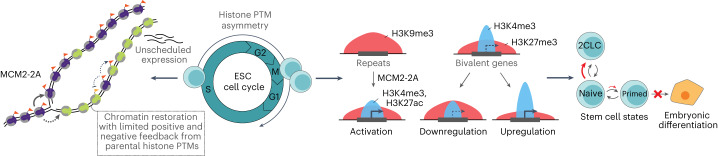
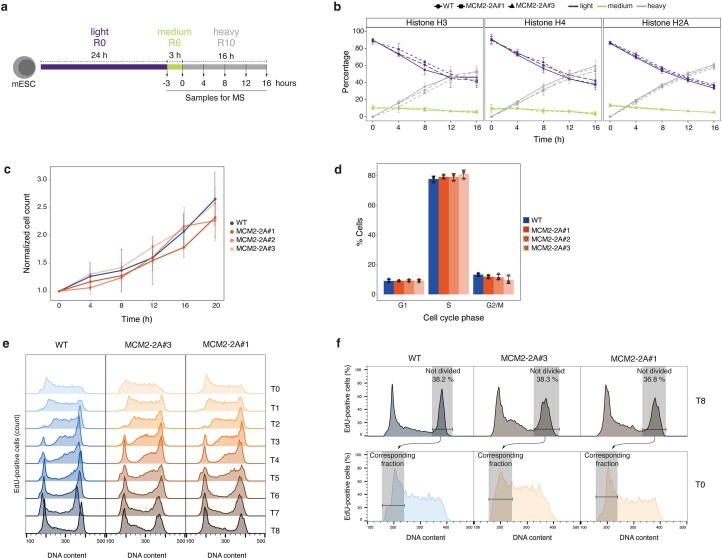
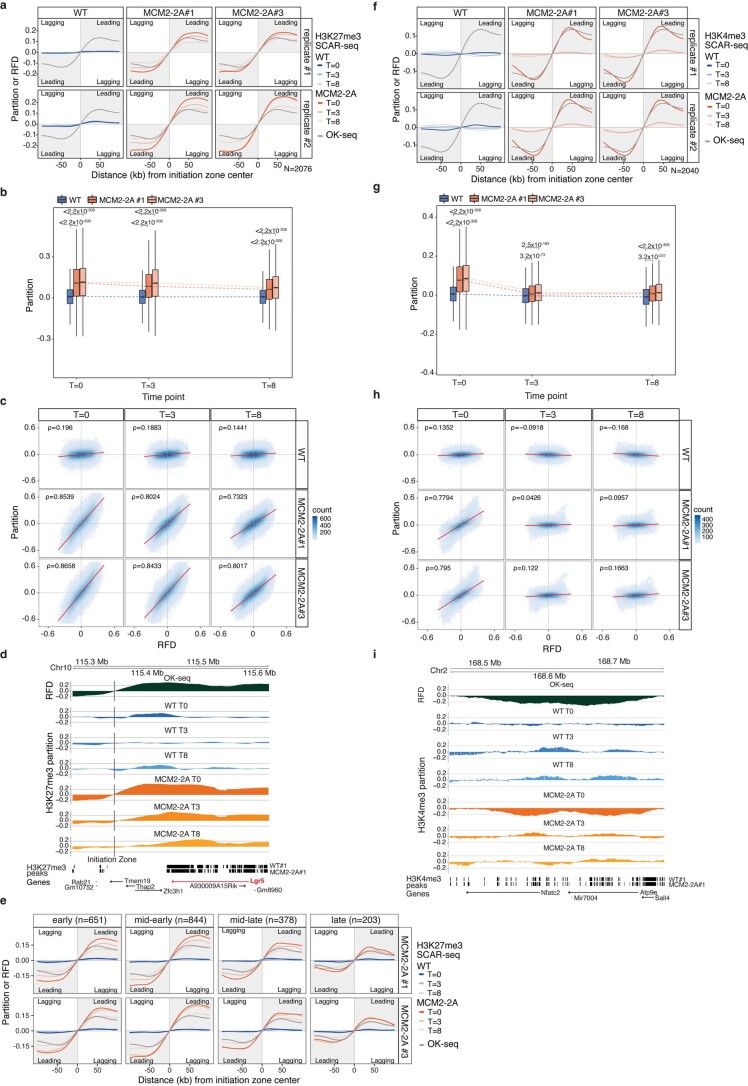
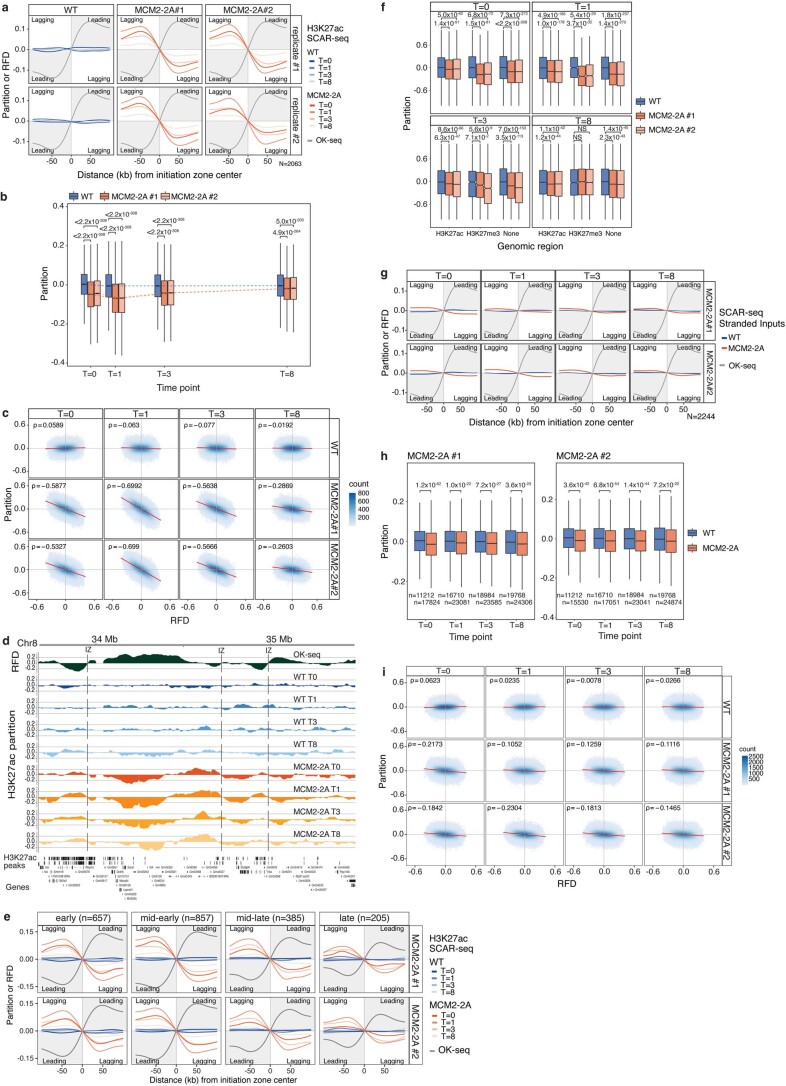
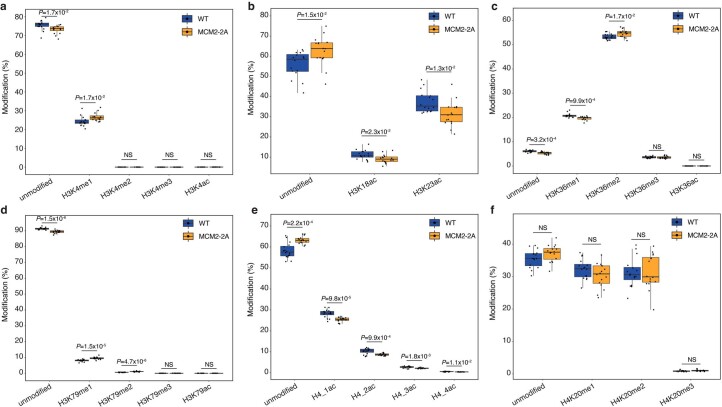
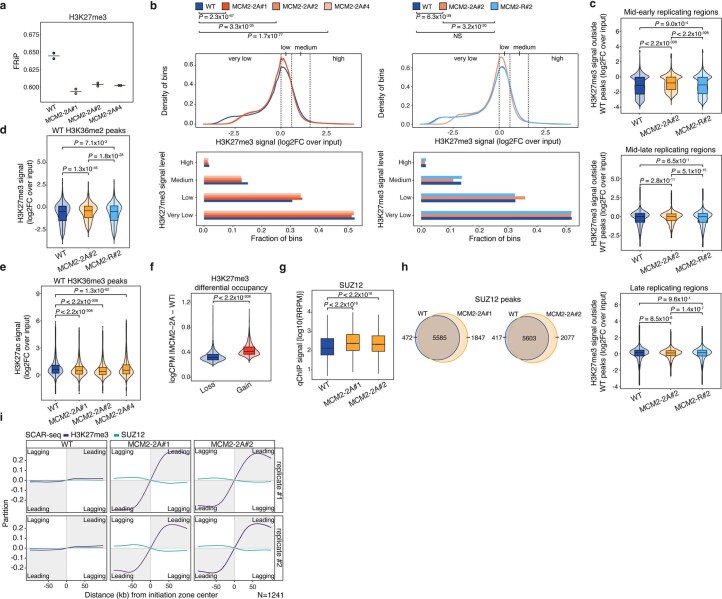
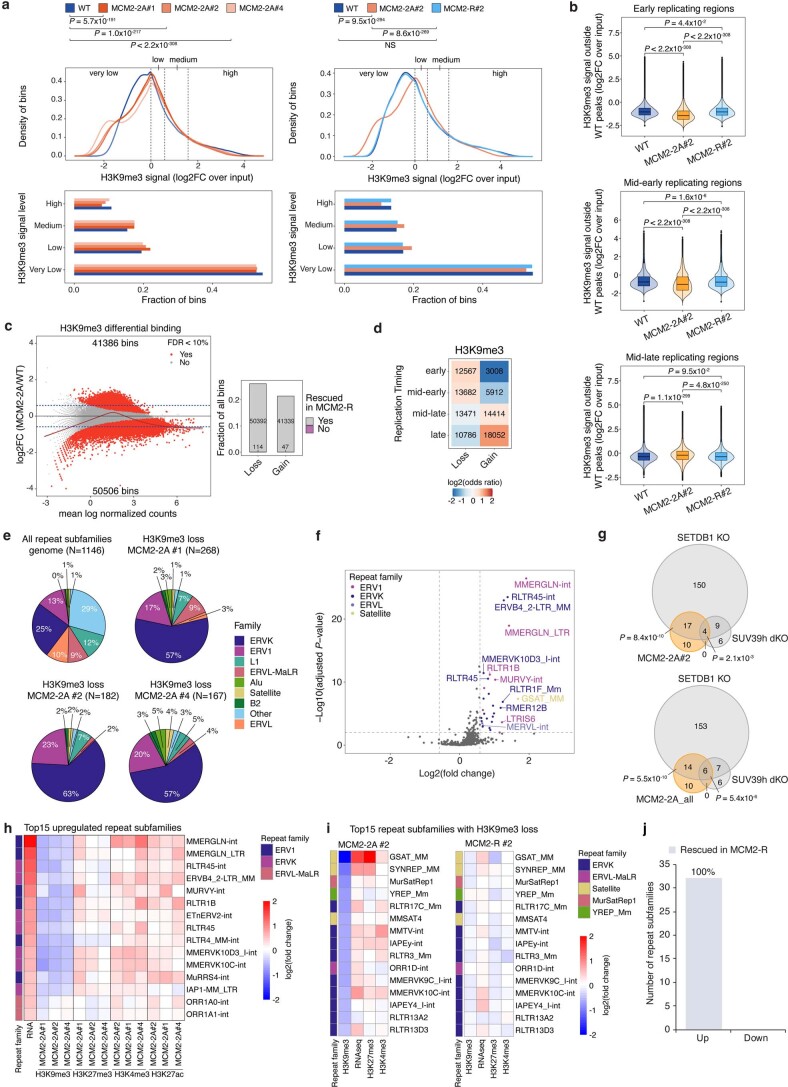
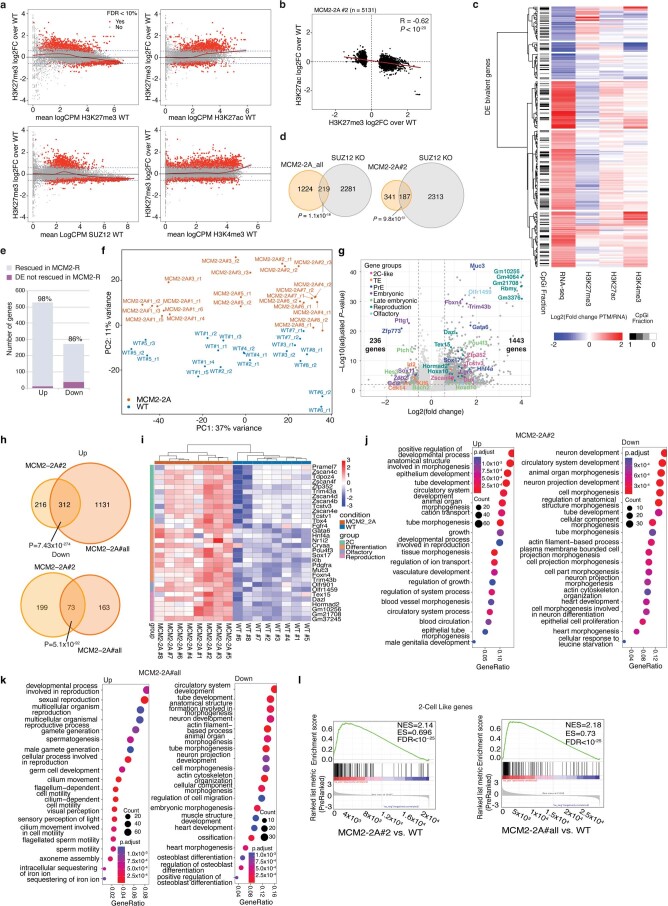
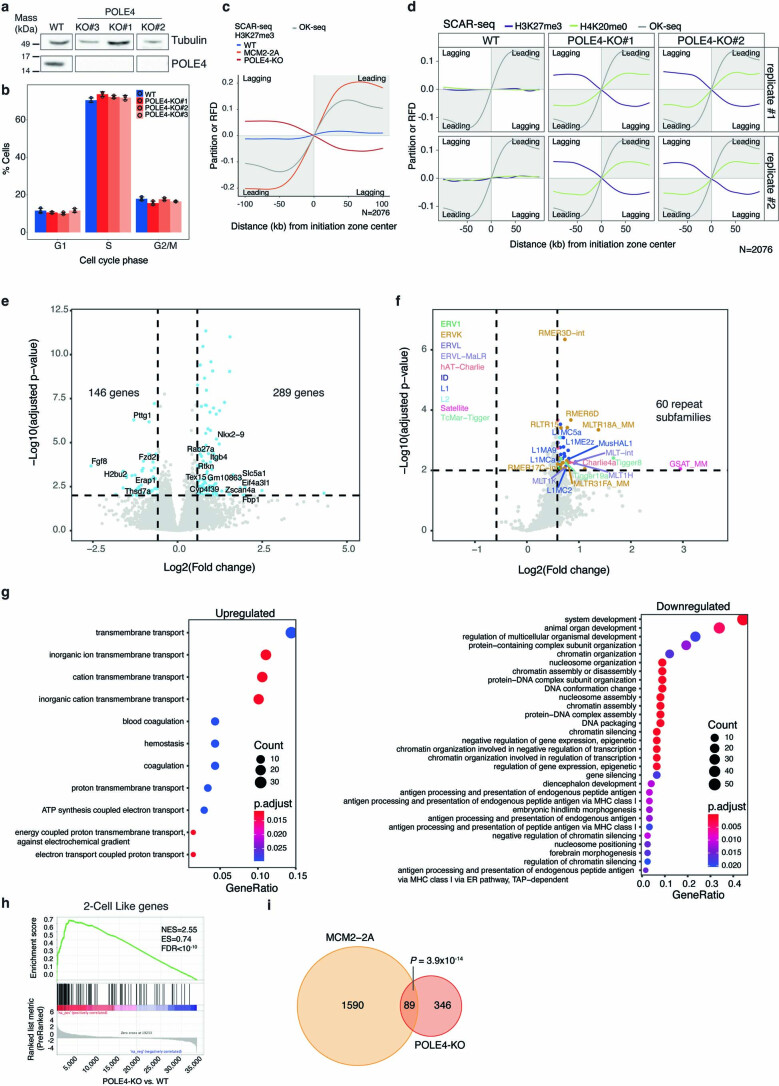
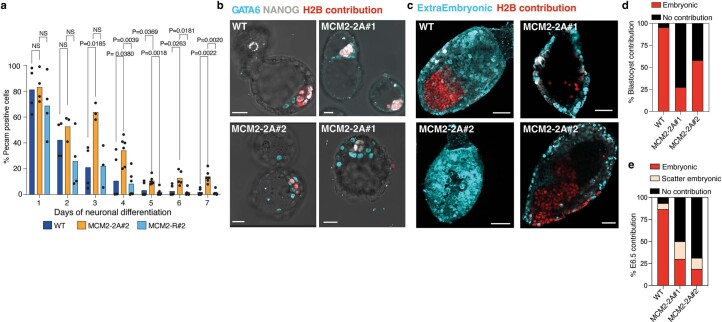
Modified parental histones are segregated symmetrically to daughter DNA strands during replication and can be inherited through mitosis. How this may sustain the epigenome and cell identity remains unknown. Here we show that transmission of histone-based information during DNA replication maintains epigenome fidelity and embryonic stem cell plasticity. Asymmetric segregation of parental histones H3-H4 in MCM2-2A mutants compromised mitotic inheritance of histone modifications and globally altered the epigenome. This included widespread spurious deposition of repressive modifications, suggesting elevated epigenetic noise. Moreover, H3K9me3 loss at repeats caused derepression and H3K27me3 redistribution across bivalent promoters correlated with misexpression of developmental genes. MCM2-2A mutation challenged dynamic transitions in cellular states across the cell cycle, enhancing naïve pluripotency and reducing lineage priming in G1. Furthermore, developmental competence was diminished, correlating with impaired exit from pluripotency. Collectively, this argues that epigenetic inheritance of histone modifications maintains a correctly balanced and dynamic chromatin landscape able to support mammalian cell differentiation.
© 2023. The Author(s).
Conflict of interest statement
A.G. is cofounder and CSO of Ankrin Therapeutics. V.S., M.V.A. and A.I. are cofounders of EpiQMAx GmbH. All other authors declare that they have no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
