

Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution
- PMID: 37666991
- PMCID: PMC10484789
- DOI: 10.1038/s41588-023-01480-1
Single-cell multi-omics identifies chronic inflammation as a driver of TP53-mutant leukemic evolution
Abstract
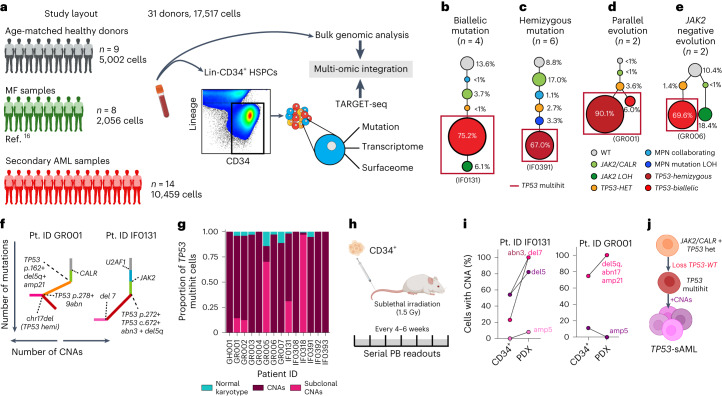
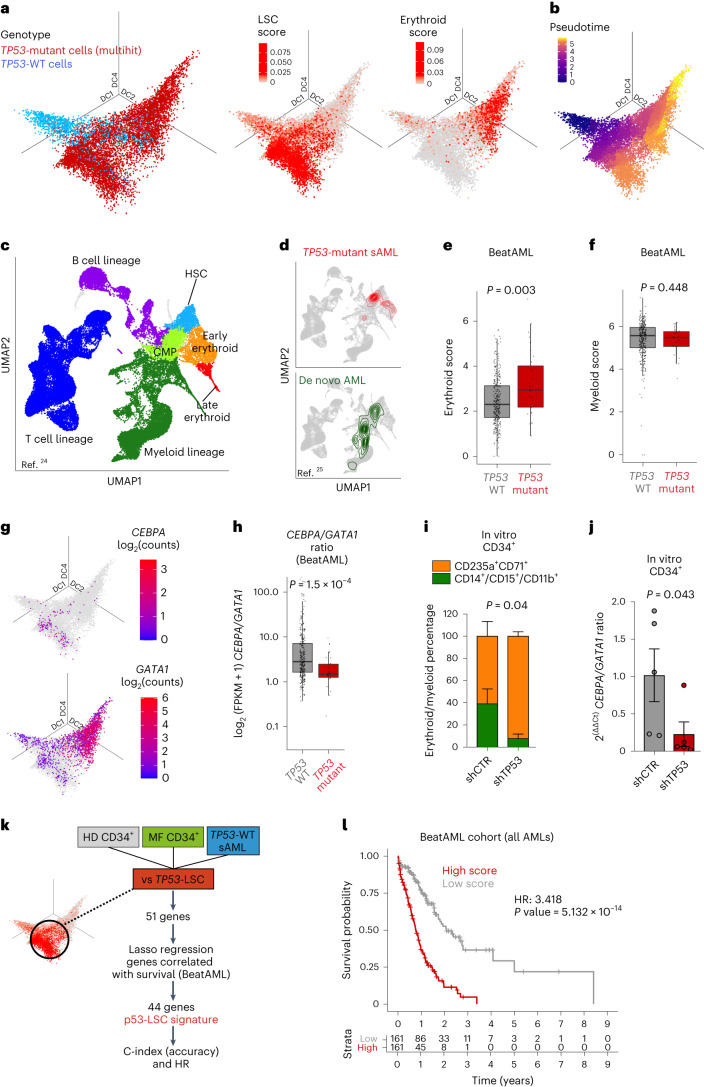
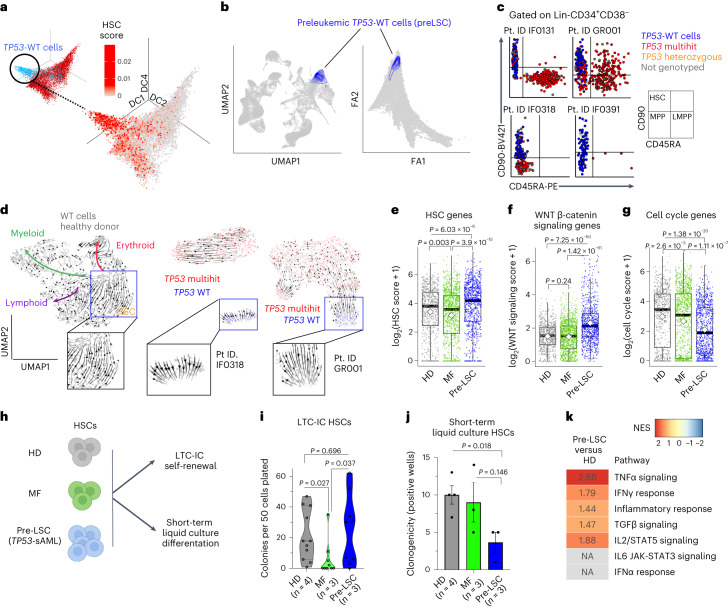
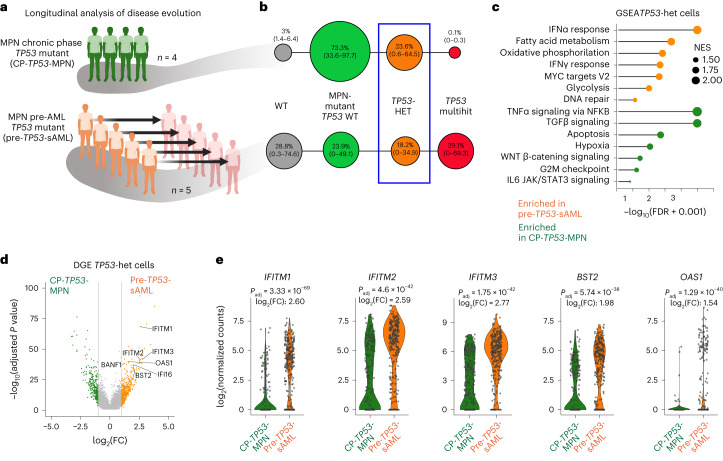
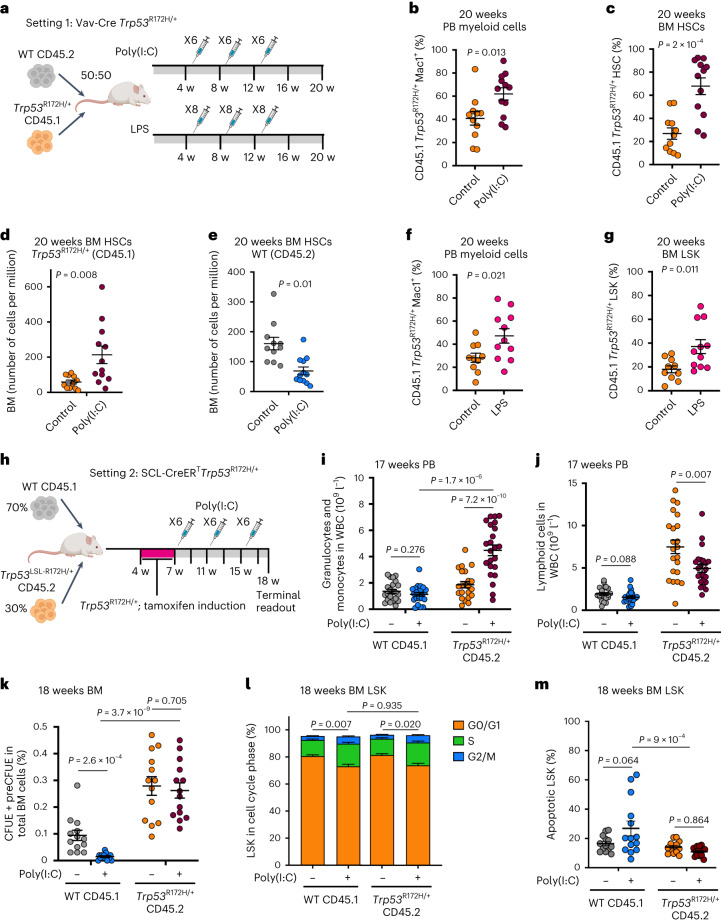
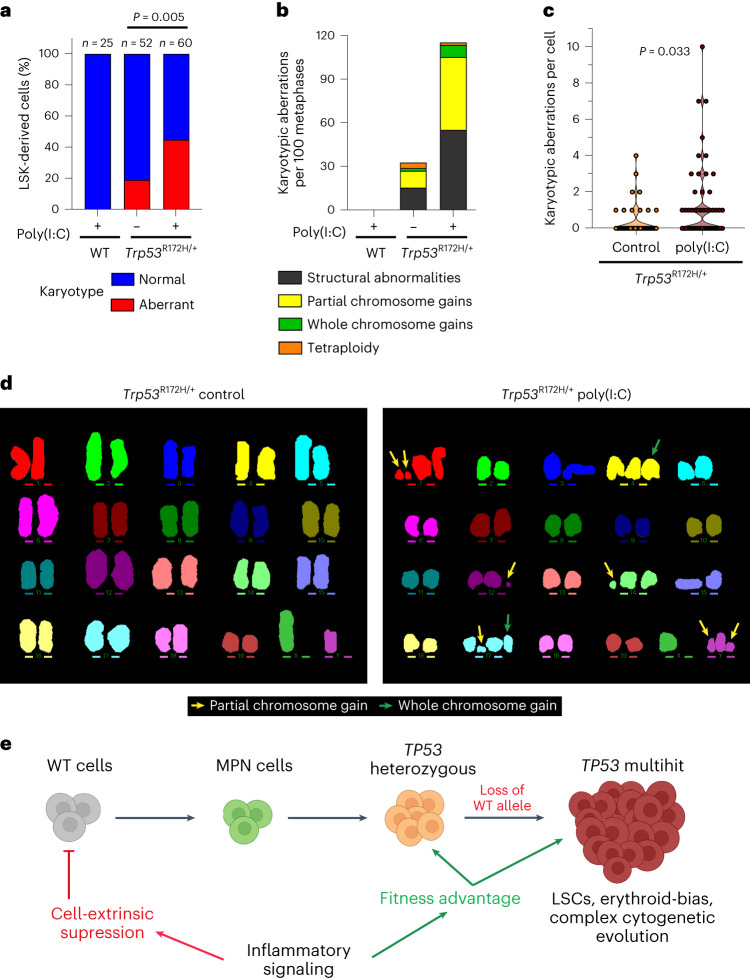
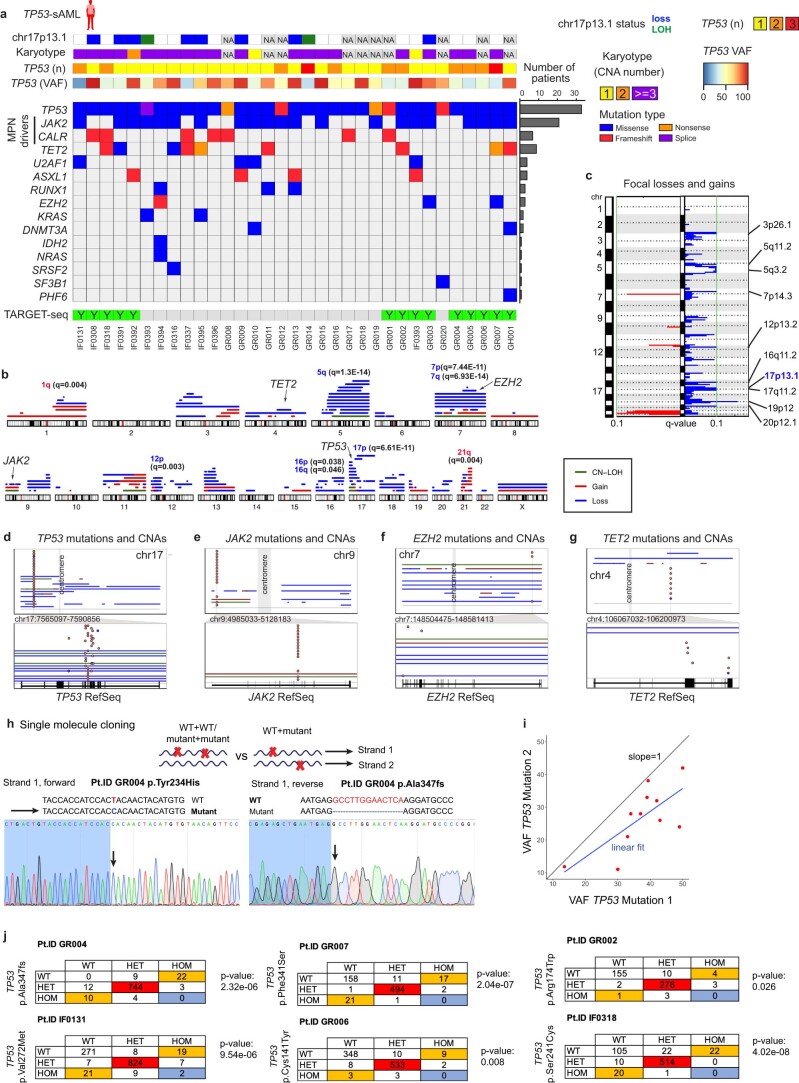
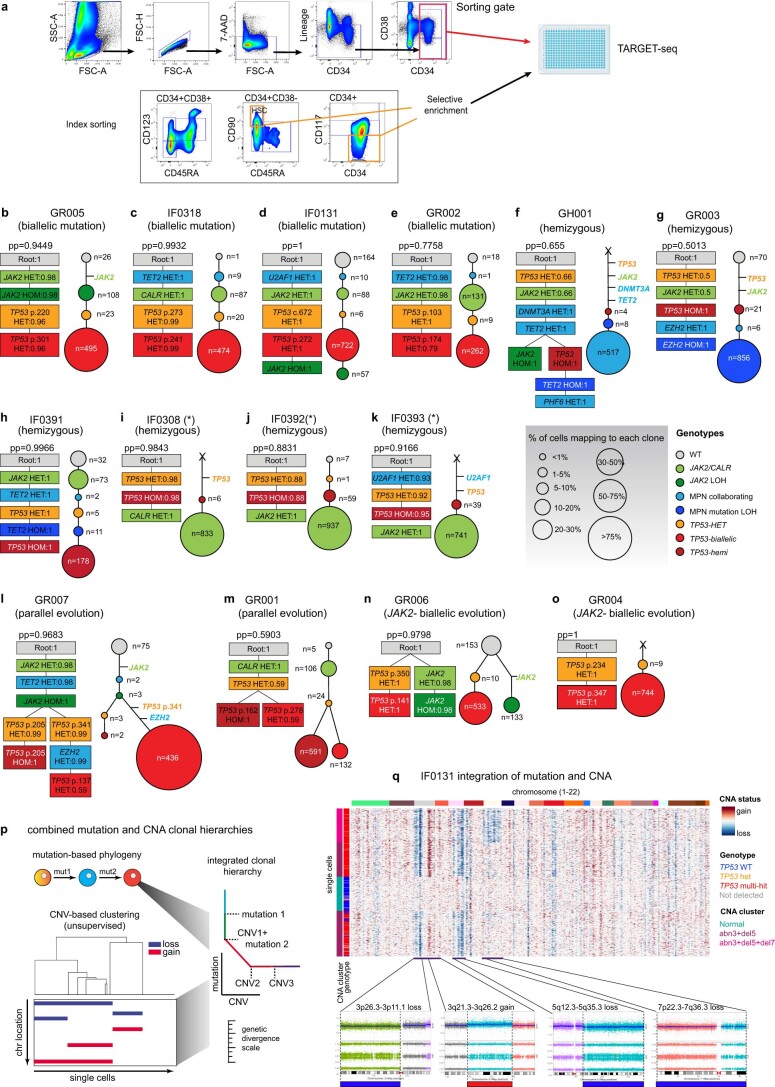
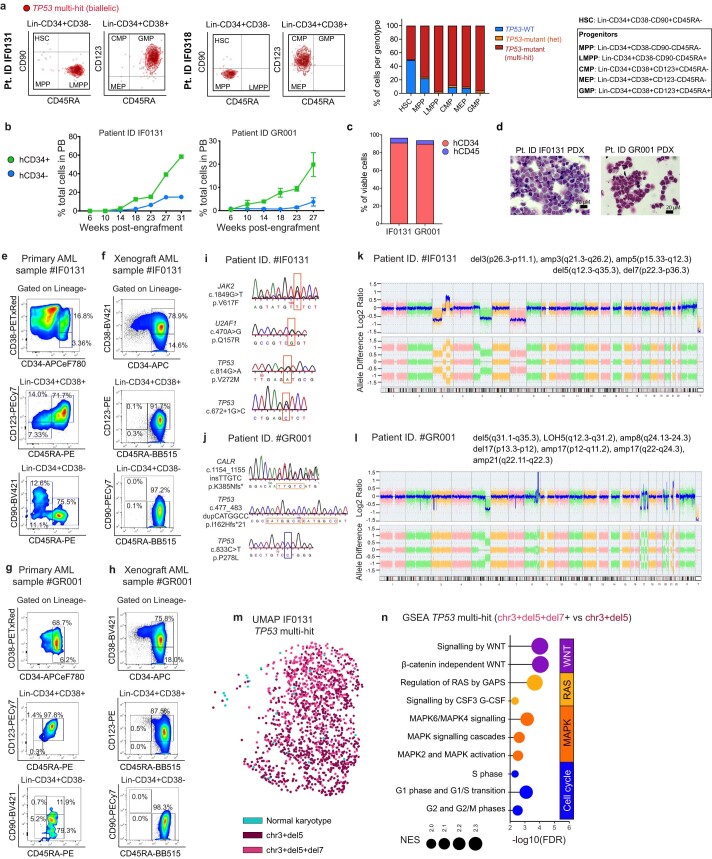
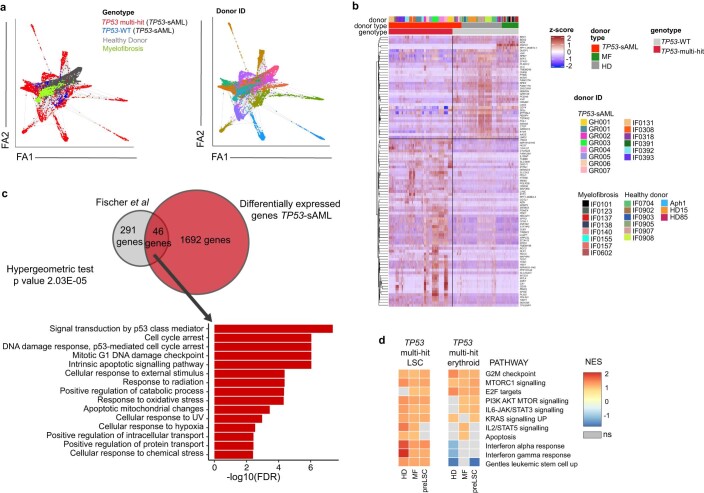
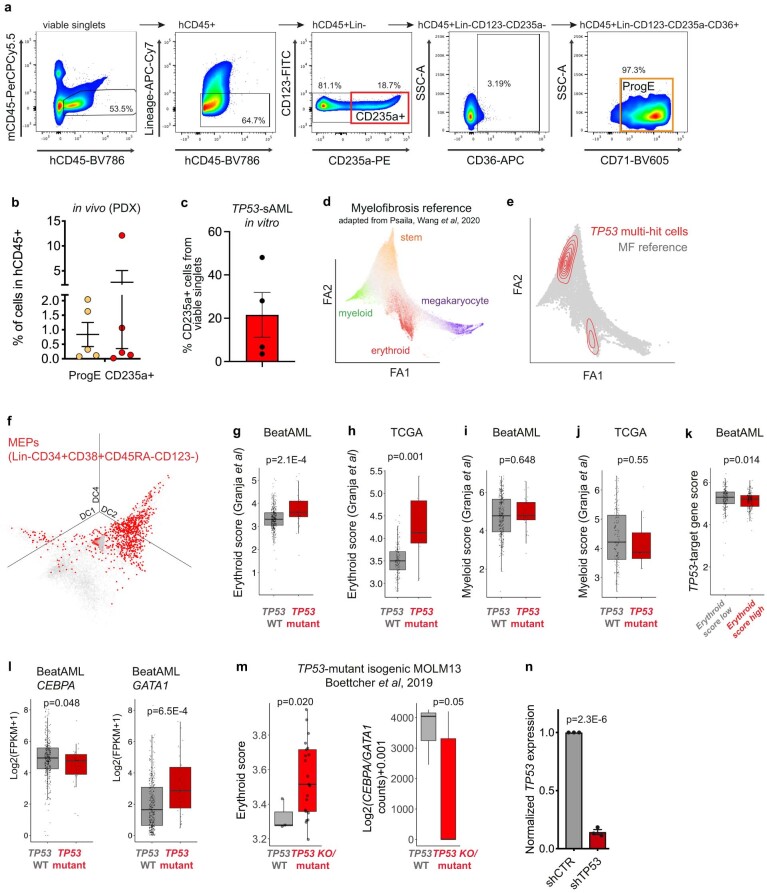
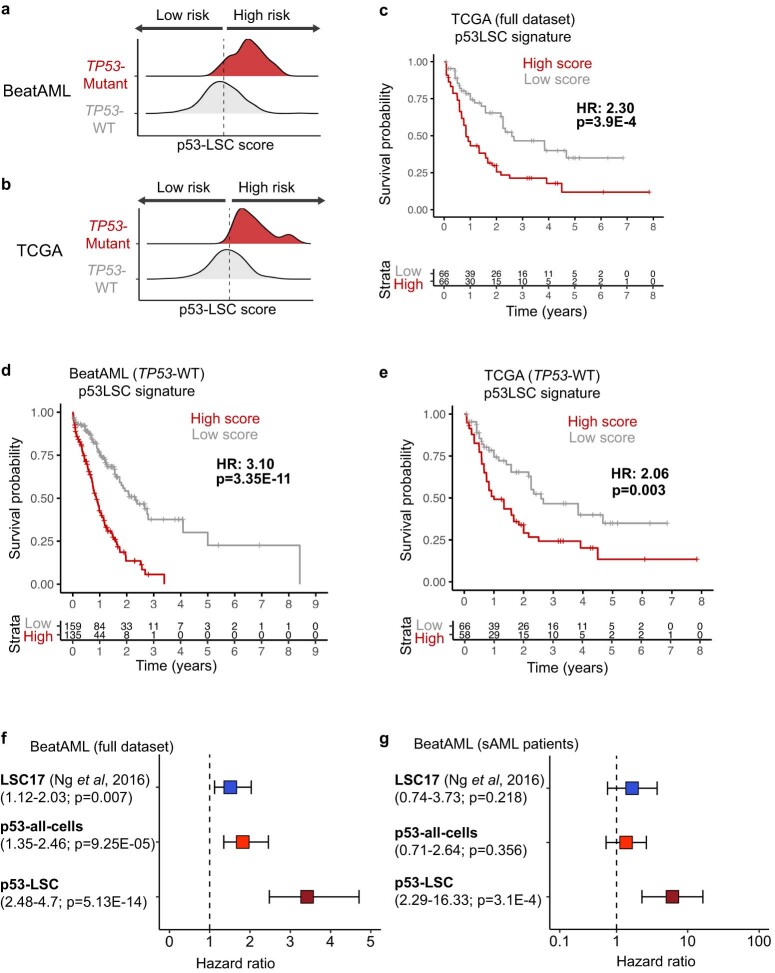
Understanding the genetic and nongenetic determinants of tumor protein 53 (TP53)-mutation-driven clonal evolution and subsequent transformation is a crucial step toward the design of rational therapeutic strategies. Here we carry out allelic resolution single-cell multi-omic analysis of hematopoietic stem/progenitor cells (HSPCs) from patients with a myeloproliferative neoplasm who transform to TP53-mutant secondary acute myeloid leukemia (sAML). All patients showed dominant TP53 'multihit' HSPC clones at transformation, with a leukemia stem cell transcriptional signature strongly predictive of adverse outcomes in independent cohorts, across both TP53-mutant and wild-type (WT) AML. Through analysis of serial samples, antecedent TP53-heterozygous clones and in vivo perturbations, we demonstrate a hitherto unrecognized effect of chronic inflammation, which suppressed TP53 WT HSPCs while enhancing the fitness advantage of TP53-mutant cells and promoted genetic evolution. Our findings will facilitate the development of risk-stratification, early detection and treatment strategies for TP53-mutant leukemia, and are of broad relevance to other cancer types.
© 2023. The Author(s).
Conflict of interest statement
A.R.M. and A.J.M. are authors on a patent related to TARGET-seq (US Patent App. 17/038,548). A patent relating to the TARGET-seq technique is licensed to Alethiomics, a spin-out company from the University of Oxford with equity owned by B.P. and A.J.M. and research funding to B.P. and A.J.M. The other authors declare no competing interests.
Figures
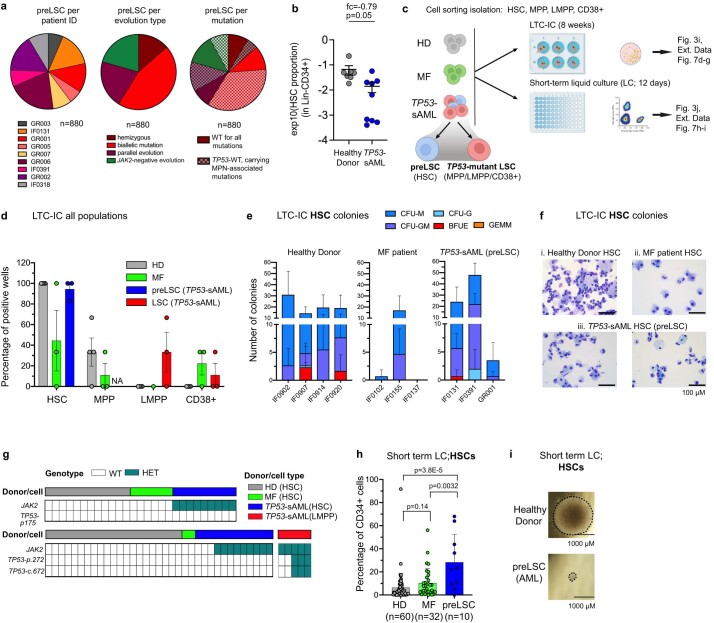
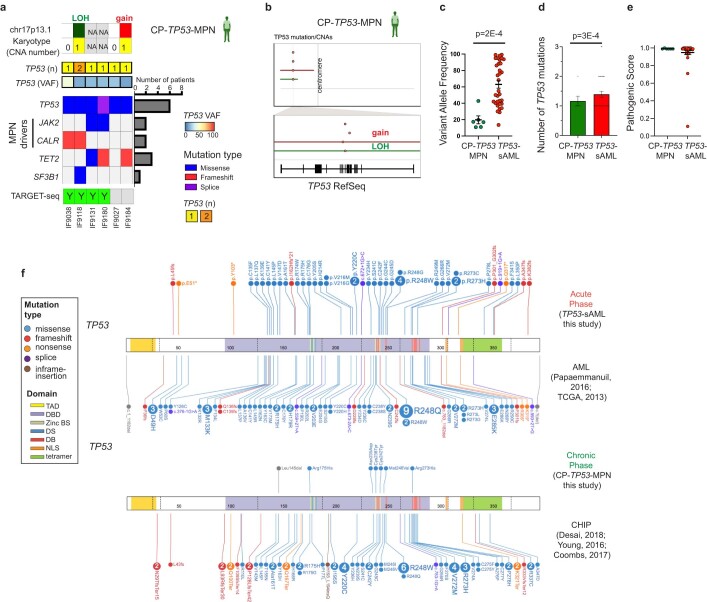
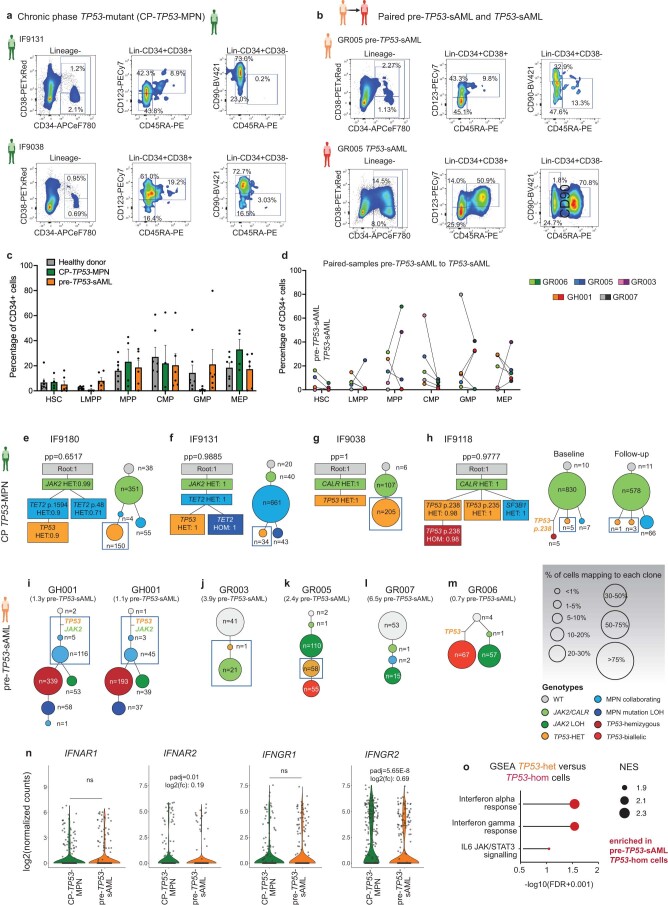
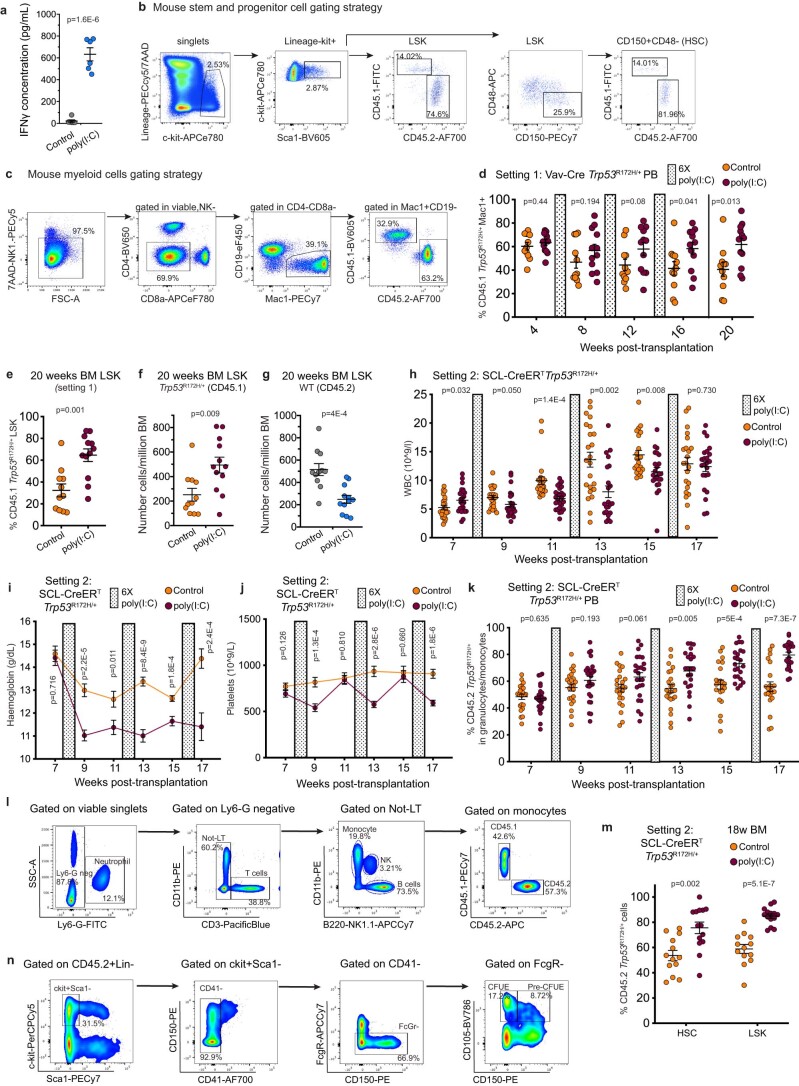
References
Publication types
MeSH terms
Substances
Grants and funding
- MR/M00919X/1/MRC_/Medical Research Council/United Kingdom
- G84/6443/MRC_/Medical Research Council/United Kingdom
- MC_PC_19049/MRC_/Medical Research Council/United Kingdom
- 28051/CRUK_/Cancer Research UK/United Kingdom
- 26988/CRUK_/Cancer Research UK/United Kingdom
- G0902418/MRC_/Medical Research Council/United Kingdom
- MC_UU_00016/5/MRC_/Medical Research Council/United Kingdom
- P30 CA008748/CA/NCI NIH HHS/United States
- 27723/CRUK_/Cancer Research UK/United Kingdom
- MR/L006340/1/MRC_/Medical Research Council/United Kingdom
- G0801073/MRC_/Medical Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- MR/R017549/1/MRC_/Medical Research Council/United Kingdom
- 27125/CRUK_/Cancer Research UK/United Kingdom
- 29034/CRUK_/Cancer Research UK/United Kingdom
- MC_UU_12009/5/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
