

A SAM analogue-utilizing ribozyme for site-specific RNA alkylation in living cells
- PMID: 37667013
- PMCID: PMC10624628
- DOI: 10.1038/s41557-023-01320-z
A SAM analogue-utilizing ribozyme for site-specific RNA alkylation in living cells
Abstract
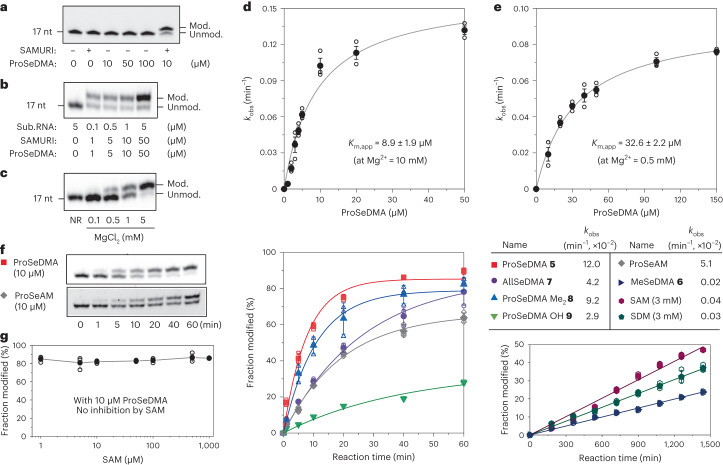
Post-transcriptional RNA modification methods are in high demand for site-specific RNA labelling and analysis of RNA functions. In vitro-selected ribozymes are attractive tools for RNA research and have the potential to overcome some of the limitations of chemoenzymatic approaches with repurposed methyltransferases. Here we report an alkyltransferase ribozyme that uses a synthetic, stabilized S-adenosylmethionine (SAM) analogue and catalyses the transfer of a propargyl group to a specific adenosine in the target RNA. Almost quantitative conversion was achieved within 1 h under a wide range of reaction conditions in vitro, including physiological magnesium ion concentrations. A genetically encoded version of the SAM analogue-utilizing ribozyme (SAMURI) was expressed in HEK293T cells, and intracellular propargylation of the target adenosine was confirmed by specific fluorescent labelling. SAMURI is a general tool for the site-specific installation of the smallest tag for azide-alkyne click chemistry, which can be further functionalized with fluorophores, affinity tags or other functional probes.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures











References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
