

Mammalian mitochondrial translation infidelity leads to oxidative stress-induced cell cycle arrest and cardiomyopathy
- PMID: 37669377
- PMCID: PMC10500172
- DOI: 10.1073/pnas.2309714120
Mammalian mitochondrial translation infidelity leads to oxidative stress-induced cell cycle arrest and cardiomyopathy
Abstract
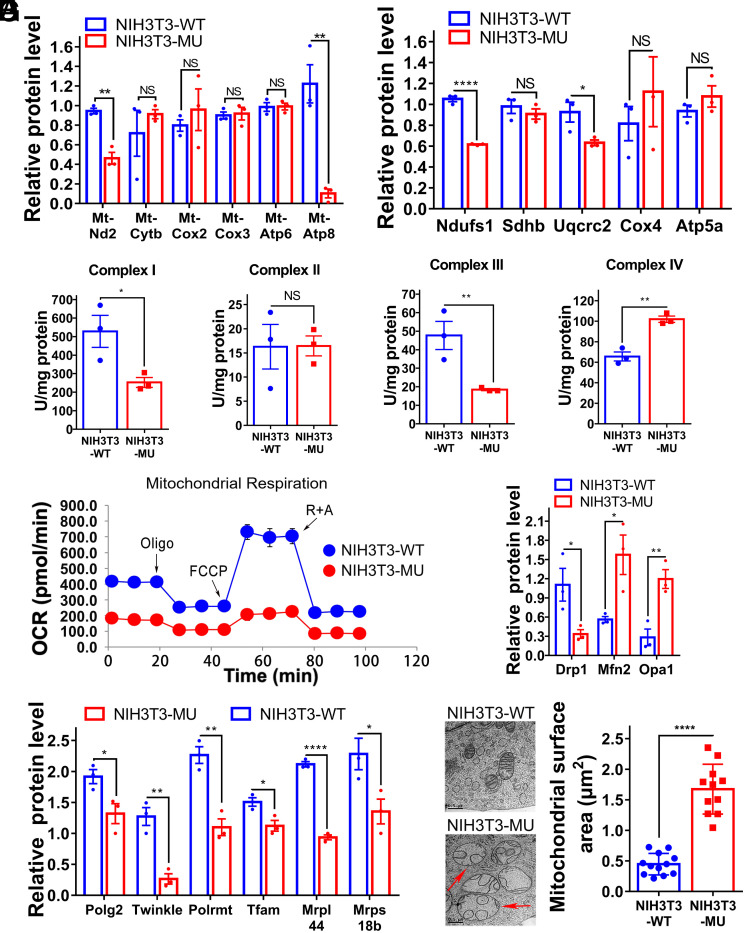
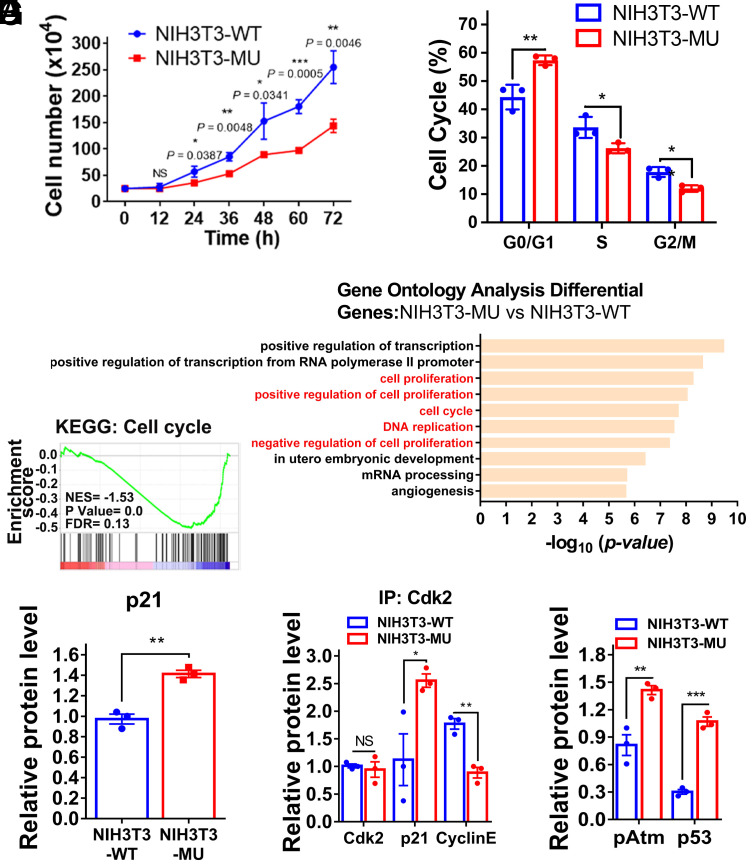
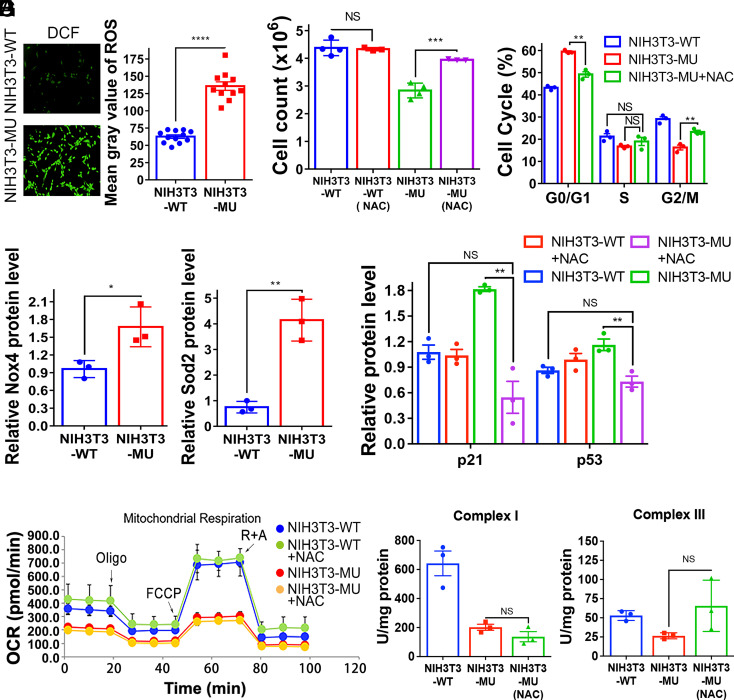
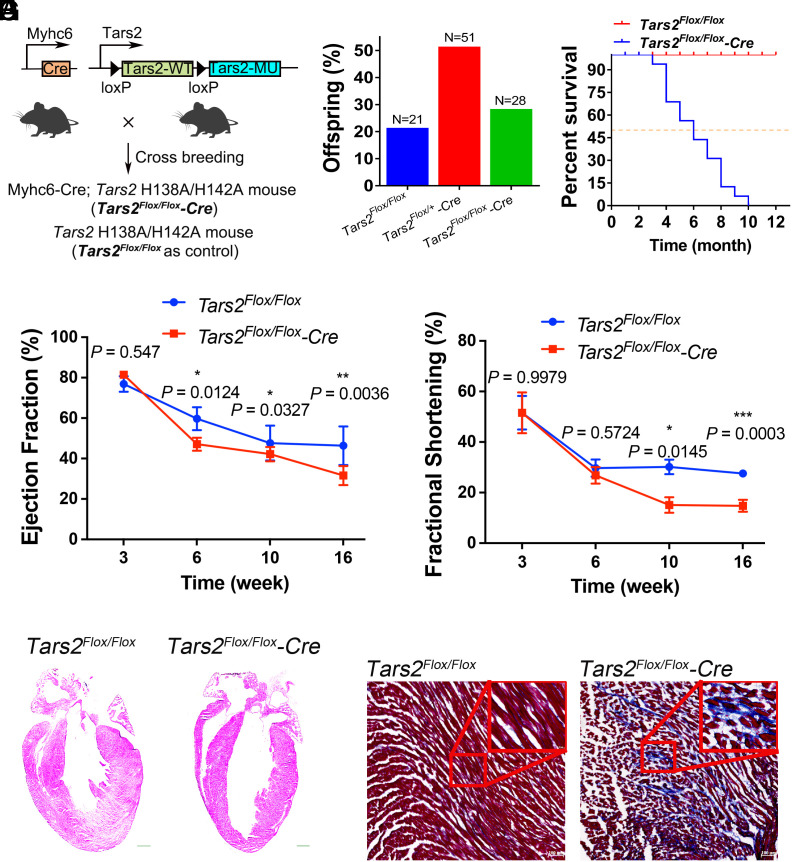
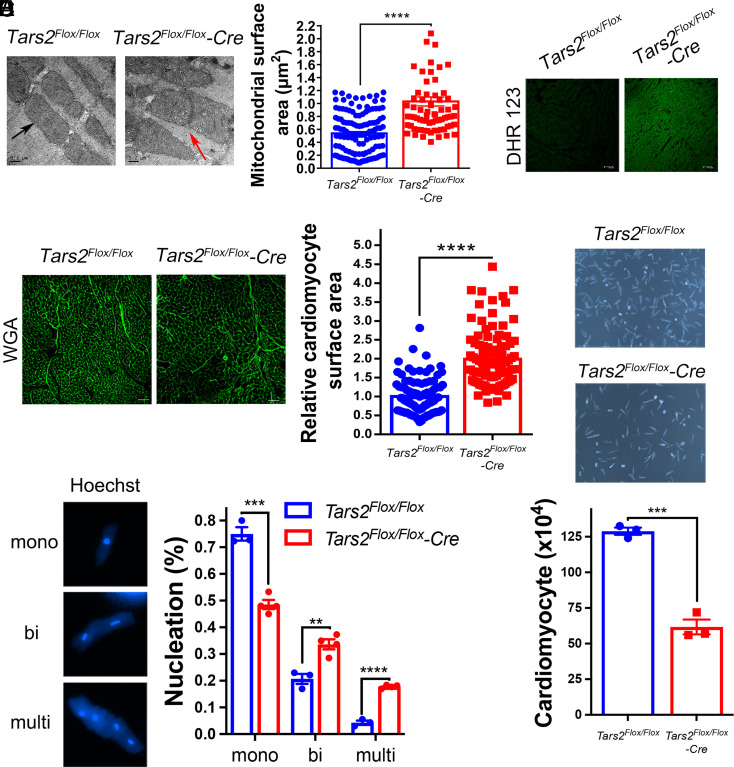
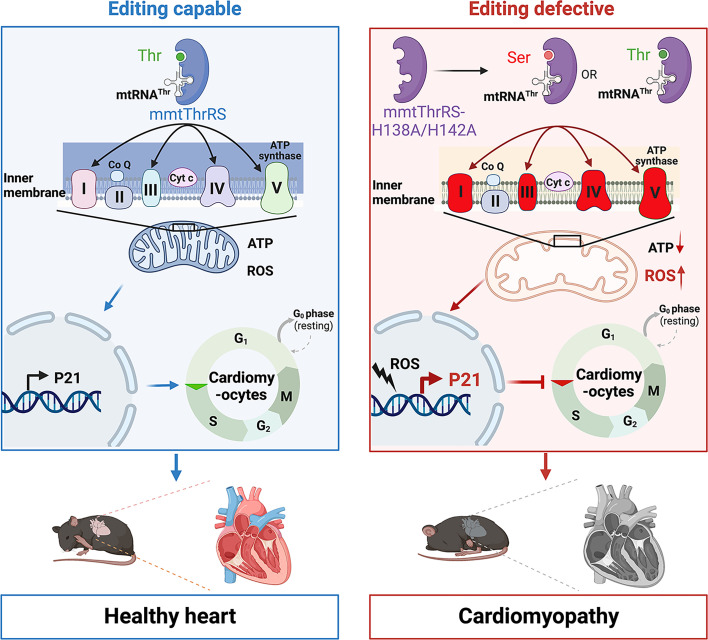
Proofreading (editing) of mischarged tRNAs by cytoplasmic aminoacyl-tRNA synthetases (aaRSs), whose impairment causes neurodegeneration and cardiac diseases, is of high significance for protein homeostasis. However, whether mitochondrial translation needs fidelity and the significance of editing by mitochondrial aaRSs have been unclear. Here, we show that mammalian cells critically depended on the editing of mitochondrial threonyl-tRNA synthetase (mtThrRS, encoded by Tars2), disruption of which accumulated Ser-tRNAThr and generated a large abundance of Thr-to-Ser misincorporated peptides in vivo. Such infidelity impaired mitochondrial translation and oxidative phosphorylation, causing oxidative stress and cell cycle arrest in the G0/G1 phase. Notably, reactive oxygen species (ROS) scavenging by N-acetylcysteine attenuated this abnormal cell proliferation. A mouse model of heart-specific defective mtThrRS editing was established. Increased ROS levels, blocked cardiomyocyte proliferation, contractile dysfunction, dilated cardiomyopathy, and cardiac fibrosis were observed. Our results elucidate that mitochondria critically require a high level of translational accuracy at Thr codons and highlight the cellular dysfunctions and imbalance in tissue homeostasis caused by mitochondrial mistranslation.
Keywords: aminoacyl-tRNA synthetase; cardiomyopathy; editing; mitochondria; tRNA.
Conflict of interest statement
The authors declare no competing interest.
Figures

Similar articles
-
Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site.Proc Natl Acad Sci U S A. 2010 Mar 2;107(9):4028-33. doi: 10.1073/pnas.1000315107. Epub 2010 Feb 16. Proc Natl Acad Sci U S A. 2010. PMID: 20160114 Free PMC article.
-
Genomic innovation of ATD alleviates mistranslation associated with multicellularity in Animalia.Elife. 2020 May 28;9:e58118. doi: 10.7554/eLife.58118. Elife. 2020. PMID: 32463355 Free PMC article.
-
Cross-editing by a tRNA synthetase allows vertebrates to abundantly express mischargeable tRNA without causing mistranslation.Nucleic Acids Res. 2020 Jul 9;48(12):6445-6457. doi: 10.1093/nar/gkaa469. Nucleic Acids Res. 2020. PMID: 32484512 Free PMC article.
-
Trans-editing by aminoacyl-tRNA synthetase-like editing domains.Enzymes. 2020;48:69-115. doi: 10.1016/bs.enz.2020.07.002. Epub 2020 Sep 8. Enzymes. 2020. PMID: 33837712 Review.
-
A review on quality control agents of protein translation - The role of Trans-editing proteins.Int J Biol Macromol. 2022 Feb 28;199:252-263. doi: 10.1016/j.ijbiomac.2021.12.176. Epub 2022 Jan 4. Int J Biol Macromol. 2022. PMID: 34995670 Review.
Cited by
-
Decoding mitochondrial stress genes in DCM: towards precision diagnosis and therapy.Hereditas. 2025 Apr 11;162(1):57. doi: 10.1186/s41065-025-00399-3. Hereditas. 2025. PMID: 40217309 Free PMC article.
-
Exploiting replication stress for synthetic lethality in MYC-driven cancers.Am J Cancer Res. 2025 Apr 15;15(4):1461-1479. doi: 10.62347/RTVX8866. eCollection 2025. Am J Cancer Res. 2025. PMID: 40371148 Free PMC article. Review.
-
Eukaryotic AlaX provides multiple checkpoints for quality and quantity of aminoacyl-tRNAs in translation.Nucleic Acids Res. 2024 Jul 22;52(13):7825-7842. doi: 10.1093/nar/gkae486. Nucleic Acids Res. 2024. PMID: 38869066 Free PMC article.
-
Multifaceted roles of t6A biogenesis in efficiency and fidelity of mitochondrial gene expression.Nucleic Acids Res. 2024 Apr 12;52(6):3213-3233. doi: 10.1093/nar/gkae013. Nucleic Acids Res. 2024. PMID: 38227555 Free PMC article.
-
Beyond protein synthesis: non-translational functions of threonyl-tRNA synthetases.Biochem Soc Trans. 2024 Apr 24;52(2):661-670. doi: 10.1042/BST20230506. Biochem Soc Trans. 2024. PMID: 38477373 Free PMC article. Review.
References
-
- Kim S., You S., Hwang D., Aminoacyl-tRNA synthetases and tumorigenesis: More than housekeeping. Nat. Rev. Cancer 11, 708–718 (2011). - PubMed
-
- Ibba M., Soll D., Quality control mechanisms during translation. Science 286, 1893–1897 (1999). - PubMed
-
- Ling J., Reynolds N., Ibba M., Aminoacyl-tRNA synthesis and translational quality control. Annu. Rev. Microbiol. 63, 61–78 (2009). - PubMed
-
- Zhou X., Wang E., Transfer RNA: A dancer between charging and mis-charging for protein biosynthesis. Sci. China Life Sci. 56, 921–932 (2013). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
