

An Autopsy Series of Seven Cases of VPS13A Disease (Chorea-Acanthocytosis)
- PMID: 37670483
- PMCID: PMC10841393
- DOI: 10.1002/mds.29589
An Autopsy Series of Seven Cases of VPS13A Disease (Chorea-Acanthocytosis)
Abstract
Background: Vacuolar protein sorting 13 homolog A (VPS13A) disease, historically known as chorea-acanthocytosis, is a rare neurodegenerative disorder caused by biallelic mutations in VPS13A, usually resulting in reduced or absent levels of its protein product, VPS13A. VPS13A localizes to contact sites between subcellular organelles, consistent with its recently identified role in lipid transfer between membranes. Mutations are associated with neuronal loss in the striatum, most prominently in the caudate nucleus, and associated marked astrogliosis. There are no other known disease-specific cellular changes (eg, protein aggregation), but autopsy reports to date have been limited, often lacking genetic or biochemical diagnostic confirmation.
Objective: The goal of this study was to characterize neuropathological findings in the brains of seven patients with VPS13A disease (chorea-acanthocytosis).
Methods: In this study, we collected brain tissues and clinical data from seven cases of VPS13A for neuropathological analysis. The clinical diagnosis was confirmed by the presence of VPS13A mutations and/or immunoblot showing the loss or reduction of VPS13A protein. Tissues underwent routine, special, and immunohistochemical staining focused on neurodegeneration. Electron microscopy was performed in one case.
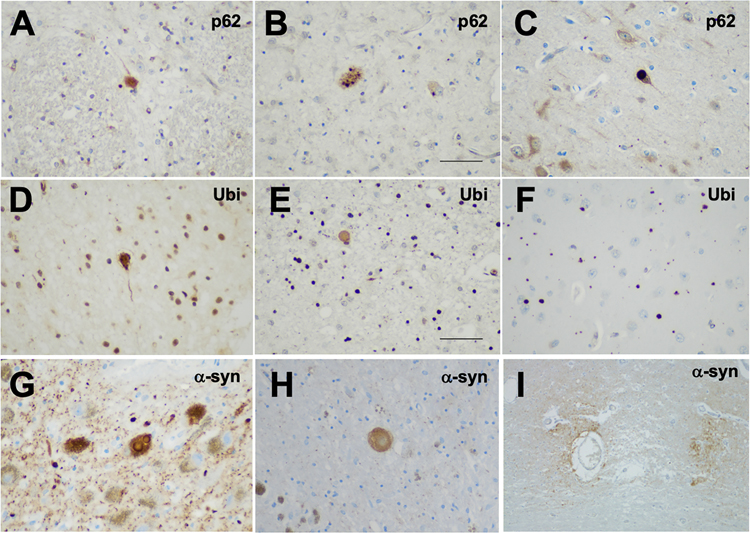
Results: Gross examination showed severe striatal atrophy. Microscopically, there was neuronal loss and astrogliosis in affected regions. Luxol fast blue staining showed variable lipid accumulation with diverse morphology, which was further characterized by electron microscopy. In some cases, rare degenerating p62- and ubiquitin-positive cells were present in affected regions. Calcifications were present in four cases, being extensive in one.
Conclusions: We present the largest autopsy series of biochemically and genetically confirmed VPS13A disease and identify novel histopathological findings implicating abnormal lipid accumulation. © 2023 International Parkinson and Movement Disorder Society.
Keywords: VPS13A; chorea-acanthocytosis; lipid; neuroacanthocytosis; neuropathology.
© 2023 International Parkinson and Movement Disorder Society.
Figures


Similar articles
-
A neuropathological study of autosomal-dominant chorea-acanthocytosis with a mutation of VPS13A.Acta Neuropathol. 2009 Jan;117(1):85-94. doi: 10.1007/s00401-008-0403-1. Epub 2008 Jun 27. Acta Neuropathol. 2009. PMID: 18584183
-
Novel heterozygous VPS13A pathogenic variants in chorea-neuroacanthocytosis: a case report.BMC Neurol. 2023 Oct 4;23(1):350. doi: 10.1186/s12883-023-03398-x. BMC Neurol. 2023. PMID: 37794323 Free PMC article.
-
Neuroacanthocytosis: a case report of chorea-acanthocytosis.J Integr Neurosci. 2019 Jun 30;18(2):197-201. doi: 10.31083/j.jin.2019.02.165. J Integr Neurosci. 2019. PMID: 31321962
-
Compound Heterozygous VPS13A Variants in a Patient with Neuroacanthocytosis: A Case Report and Review of the Literature.Lab Med. 2022 Jul 4;53(4):433-435. doi: 10.1093/labmed/lmab124. Lab Med. 2022. PMID: 35075478 Review.
-
Two case reports of chorea-acanthocytosis and review of literature.Eur J Med Res. 2022 Feb 7;27(1):22. doi: 10.1186/s40001-022-00646-7. Eur J Med Res. 2022. PMID: 35130982 Free PMC article. Review.
Cited by
-
Multiple sclerosis in LRRK2 G2019S Parkinson's disease and isolated nigral degeneration in a homozygous variant carrier.Front Neurol. 2024 Aug 8;15:1450654. doi: 10.3389/fneur.2024.1450654. eCollection 2024. Front Neurol. 2024. PMID: 39175765 Free PMC article.
-
GATA Transcription Factors: A Cross-Road for Erythropoiesis, Neurodevelopment, and Synucleinopathies.Dev Neurobiol. 2025 Jul;85(3):e22975. doi: 10.1002/dneu.22975. Dev Neurobiol. 2025. PMID: 40474753 Free PMC article. Review.
-
Neuroacanthocytosis Syndromes: The Clinical Perspective.Contact (Thousand Oaks). 2023 Dec 10;6:25152564231210339. doi: 10.1177/25152564231210339. eCollection 2023 Jan-Dec. Contact (Thousand Oaks). 2023. PMID: 38090146 Free PMC article. Review.
-
Neurodegenerative and Neurodevelopmental Roles for Bulk Lipid Transporters VPS13A and BLTP2.Mov Disord. 2025 Jul;40(7):1356-1368. doi: 10.1002/mds.30178. Epub 2025 Mar 28. Mov Disord. 2025. PMID: 40152532 Free PMC article.
-
Analysis of Brain, Blood, and Testis Phenotypes Lacking the Vps13a Gene in C57BL/6N Mice.Int J Mol Sci. 2024 Jul 16;25(14):7776. doi: 10.3390/ijms25147776. Int J Mol Sci. 2024. PMID: 39063018 Free PMC article.
References
-
- Peikert K et al. VPS13A Disease. in GeneReviews® (eds. Adam MP et al.) (University of Washington, Seattle, 1993). - PubMed
-
- Rampoldi L et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat. Genet. 28, 119–120 (2001). - PubMed
-
- Ueno S et al. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat. Genet. 28, 121–122 (2001). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 AG063819/AG/NIA NIH HHS/United States
- R01NS116006/NH/NIH HHS/United States
- R01 AG062348/AG/NIA NIH HHS/United States
- P30 AG072978/AG/NIA NIH HHS/United States
- R01AG062348/NH/NIH HHS/United States
- R01NS095252/NH/NIH HHS/United States
- R01 NS086736/NS/NINDS NIH HHS/United States
- R01 NS095252/NS/NINDS NIH HHS/United States
- U54NS115266/NH/NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01AG054008/NH/NIH HHS/United States
- R21 NS127037/NS/NINDS NIH HHS/United States
- RF1 NS095252/NS/NINDS NIH HHS/United States
- RF1 MH128969/MH/NIMH NIH HHS/United States
- RF1AG060961/NH/NIH HHS/United States
- U54 NS115266/NS/NINDS NIH HHS/United States
- RF1MH128969/NH/NIH HHS/United States
- R01 NS116006/NS/NINDS NIH HHS/United States
- R01NS086736/NH/NIH HHS/United States
- RF1 AG062348/AG/NIA NIH HHS/United States
- RF1 AG060961/AG/NIA NIH HHS/United States
- P30AG066514/NH/NIH HHS/United States
- P30 AG072959/AG/NIA NIH HHS/United States
- P30 AG066514/AG/NIA NIH HHS/United States
- R01AG063819/NH/NIH HHS/United States
- R01 AG054008/AG/NIA NIH HHS/United States
- T32 AG052909/AG/NIA NIH HHS/United States
- I01 CX002342/CX/CSRD VA/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
