

Automated machine learning for genome wide association studies
- PMID: 37672022
- PMCID: PMC10562960
- DOI: 10.1093/bioinformatics/btad545
Automated machine learning for genome wide association studies
Abstract
Motivation: Genome-wide association studies (GWAS) present several computational and statistical challenges for their data analysis, including knowledge discovery, interpretability, and translation to clinical practice.
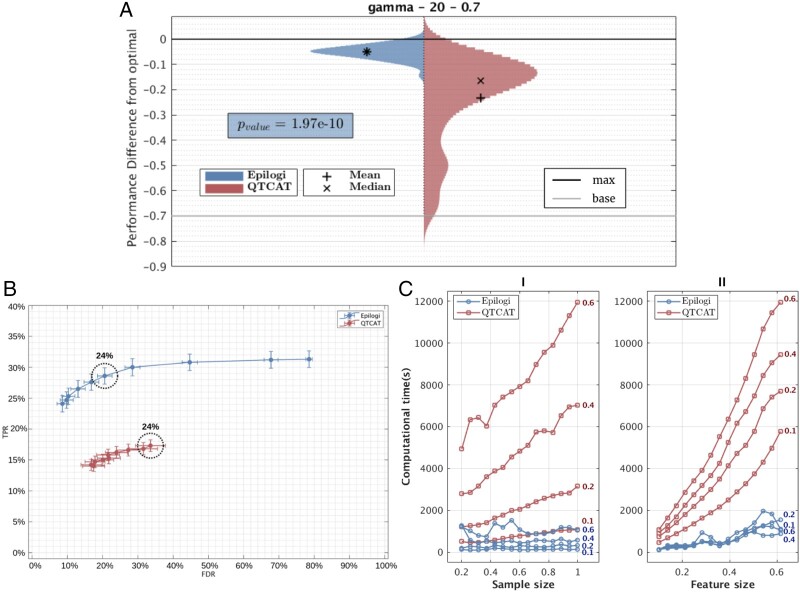
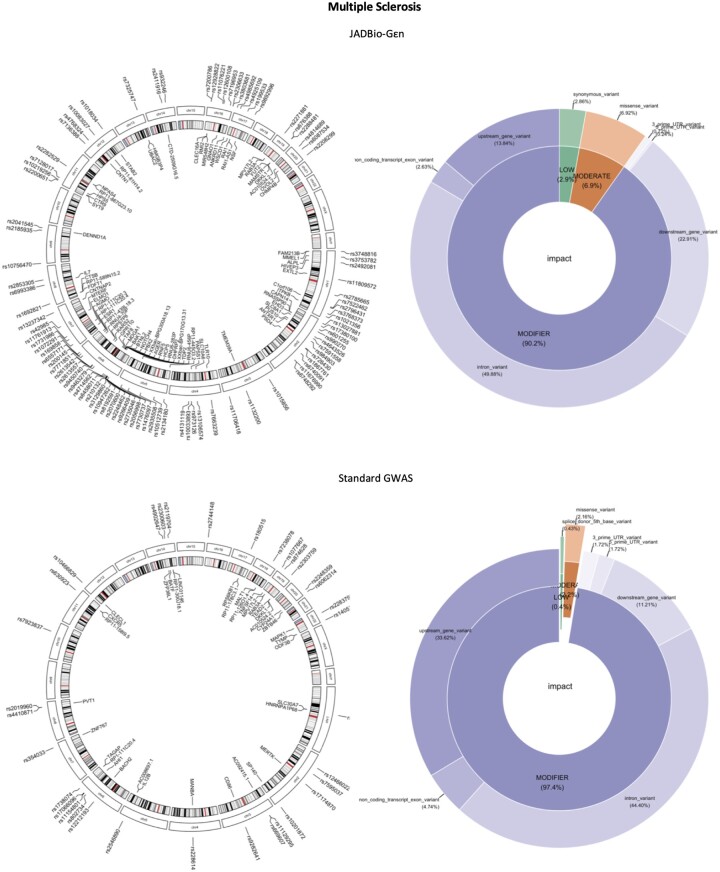
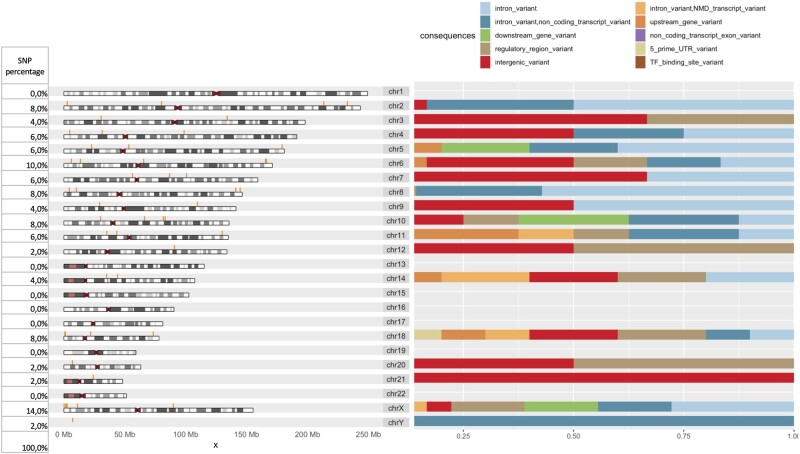
Results: We develop, apply, and comparatively evaluate an automated machine learning (AutoML) approach, customized for genomic data that delivers reliable predictive and diagnostic models, the set of genetic variants that are important for predictions (called a biosignature), and an estimate of the out-of-sample predictive power. This AutoML approach discovers variants with higher predictive performance compared to standard GWAS methods, computes an individual risk prediction score, generalizes to new, unseen data, is shown to better differentiate causal variants from other highly correlated variants, and enhances knowledge discovery and interpretability by reporting multiple equivalent biosignatures.
Availability and implementation: Code for this study is available at: https://github.com/mensxmachina/autoML-GWAS. JADBio offers a free version at: https://jadbio.com/sign-up/. SNP data can be downloaded from the EGA repository (https://ega-archive.org/). PRS data are found at: https://www.aicrowd.com/challenges/opensnp-height-prediction. Simulation data to study population structure can be found at: https://easygwas.ethz.ch/data/public/dataset/view/1/.
© The Author(s) 2023. Published by Oxford University Press.
Conflict of interest statement
I.T., P.C., Z.P., S.F., and V.L. are or were directly or indirectly affiliated with Gnosis Data Analysis that offers the JADBio service commercially.
Figures
References
-
- Adamou M, Antoniou G, Greasidou E. et al. Toward automatic risk assessment to support suicide prevention. Crisis 2019;40:249–56. - PubMed
-
- Agrapetidou A, Charonyktakis P, Gogas P. et al. An AutoML application to forecasting bank failures. Appl Econ Lett 2021;28:5–9.
-
- Batsakis S, Adamou M, Tachmazidis I. et al. Data-driven decision support for autism diagnosis using machine learning. Digital 2022;2:224.
-
- Borboudakis G, Stergiannakos T, Frysali M. et al. Chemically intuited, large-scale screening of MOFs by machine learning techniques. npj Comput Mater 2017;3:40.
