

Hydrogen-bond-acceptor ligands enable distal C(sp3)-H arylation of free alcohols
- PMID: 37674074
- PMCID: PMC11139439
- DOI: 10.1038/s41586-023-06485-8
Hydrogen-bond-acceptor ligands enable distal C(sp3)-H arylation of free alcohols
Abstract
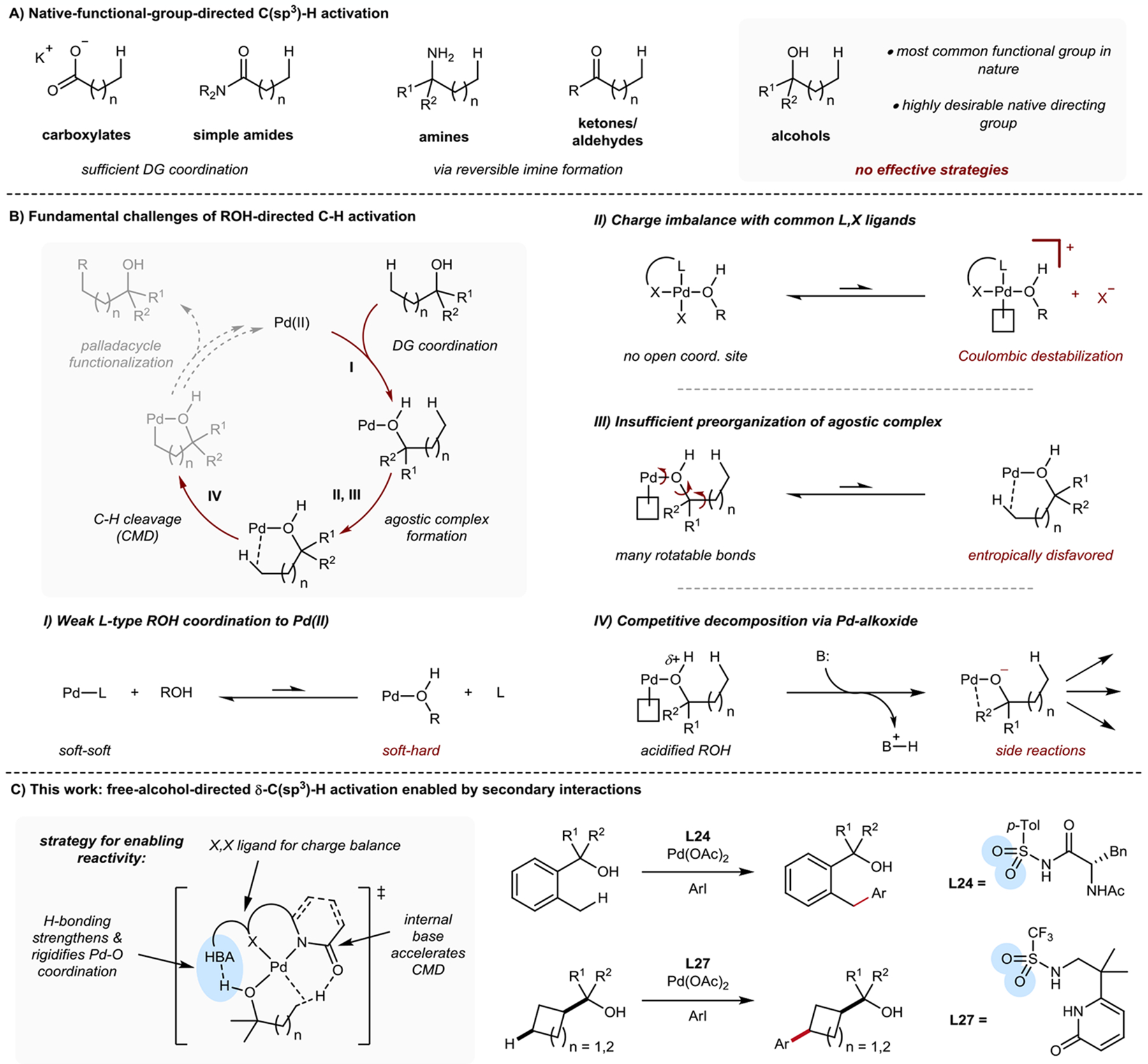
The functionalization of C-H bonds in organic molecules is one of the most direct approaches for chemical synthesis. Recent advances in catalysis have allowed native chemical groups such as carboxylic acids, ketones and amines to control and direct C(sp3)-H activation1-4. However, alcohols, among the most common functionalities in organic chemistry5, have remained intractable because of their low affinity for late transition-metal catalysts6,7. Here we describe ligands that enable alcohol-directed arylation of δ-C(sp3)-H bonds. We use charge balance and a secondary-coordination-sphere hydrogen-bonding interaction-evidenced by structure-activity relationship studies, computational modelling and crystallographic data-to stabilize L-type hydroxyl coordination to palladium, thereby facilitating the assembly of the key C-H cleavage transition state. In contrast to previous studies in C-H activation, in which secondary interactions were used to control selectivity in the context of established reactivity8-13, this report demonstrates the feasibility of using secondary interactions to enable challenging, previously unknown reactivity by enhancing substrate-catalyst affinity.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures







References
-
- Chen Z, Wang B, Zhang J, Yu W, Liu Z & Zhang Y Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups. Org. Chem. Front 2, 1107–1295 (2015).
-
- Uttry A & van Gemmeren M Direct C(sp3)–H activation of carboxylic acids. Synthesis 52, 479–488 (2020).
-
- Higham JI & Bull JA Transient imine directing groups for the C–H functionalization of aldehydes, ketones and amines: an update 2018–2020. Org. Biomol. Chem 18, 7291–7315 (2020). - PubMed
-
- Ertl P & Schuhmann T A systematic cheminformatics analysis of functional groups occurring in natural products. J. Nat. Prod 82, 1258–1263 (2019). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
