

Pervasive downstream RNA hairpins dynamically dictate start-codon selection
- PMID: 37674078
- PMCID: PMC10499604
- DOI: 10.1038/s41586-023-06500-y
Pervasive downstream RNA hairpins dynamically dictate start-codon selection
Abstract
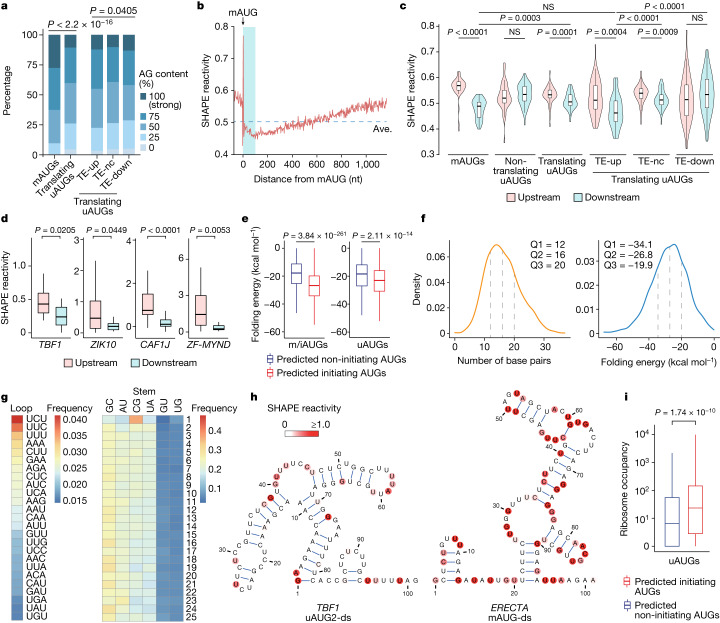
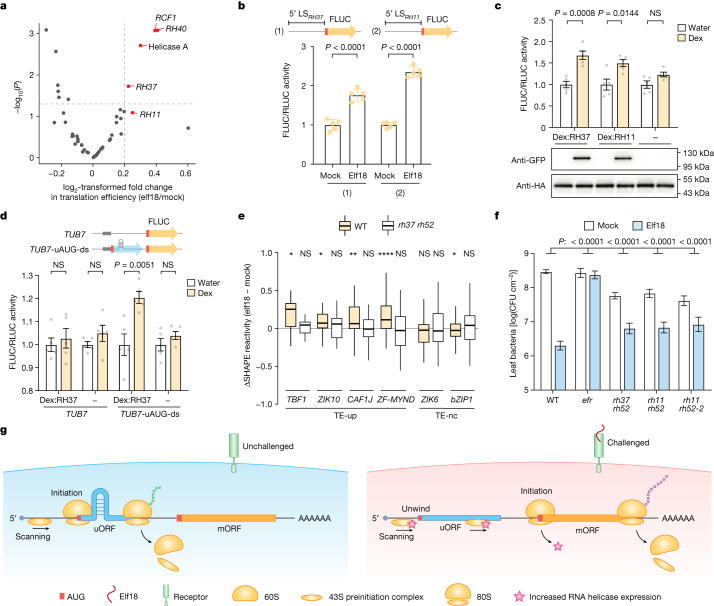
Translational reprogramming allows organisms to adapt to changing conditions. Upstream start codons (uAUGs), which are prevalently present in mRNAs, have crucial roles in regulating translation by providing alternative translation start sites1-4. However, what determines this selective initiation of translation between conditions remains unclear. Here, by integrating transcriptome-wide translational and structural analyses during pattern-triggered immunity in Arabidopsis, we found that transcripts with immune-induced translation are enriched with upstream open reading frames (uORFs). Without infection, these uORFs are selectively translated owing to hairpins immediately downstream of uAUGs, presumably by slowing and engaging the scanning preinitiation complex. Modelling using deep learning provides unbiased support for these recognizable double-stranded RNA structures downstream of uAUGs (which we term uAUG-ds) being responsible for the selective translation of uAUGs, and allows the prediction and rational design of translating uAUG-ds. We found that uAUG-ds-mediated regulation can be generalized to human cells. Moreover, uAUG-ds-mediated start-codon selection is dynamically regulated. After immune challenge in plants, induced RNA helicases that are homologous to Ded1p in yeast and DDX3X in humans resolve these structures, allowing ribosomes to bypass uAUGs to translate downstream defence proteins. This study shows that mRNA structures dynamically regulate start-codon selection. The prevalence of this RNA structural feature and the conservation of RNA helicases across kingdoms suggest that mRNA structural remodelling is a general feature of translational reprogramming.
© 2023. The Author(s).
Conflict of interest statement
X.D. is a founder of Upstream Biotechnology and a member of its scientific advisory board, as well as a member of the scientific advisory board of Inari Agriculture and Aferna Bio. K.M.W. is an advisor to and holds equity in Ribometrix. X.D. and Y.X. are listed as co-inventors on a patent application (no. 63/432,775) related to this work. The remaining other authors declare no competing interests.
Figures












Comment in
-
Dynamic regulation of messenger RNA structure controls translation.Nature. 2023 Sep;621(7978):259-260. doi: 10.1038/d41586-023-02673-8. Nature. 2023. PMID: 37674086 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
