

Modeling of African population history using f-statistics is biased when applying all previously proposed SNP ascertainment schemes
- PMID: 37676865
- PMCID: PMC10508636
- DOI: 10.1371/journal.pgen.1010931
Modeling of African population history using f-statistics is biased when applying all previously proposed SNP ascertainment schemes
Abstract
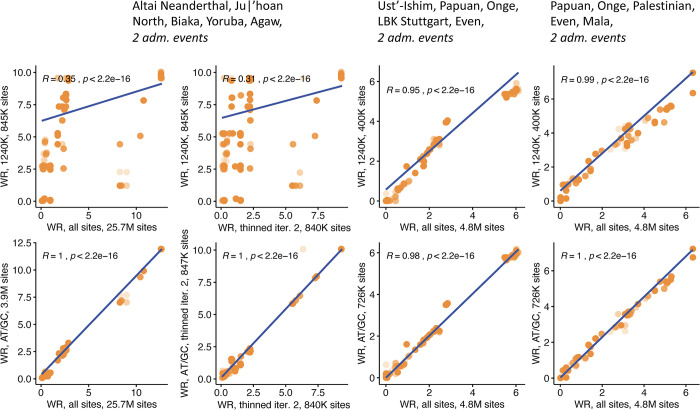
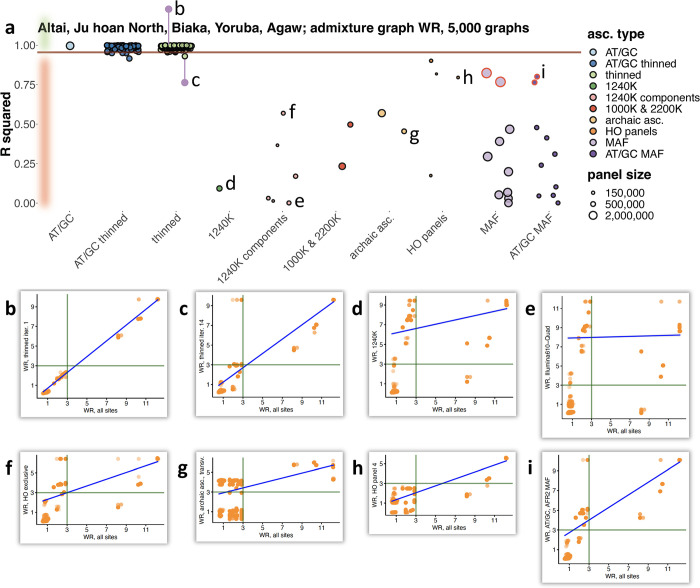
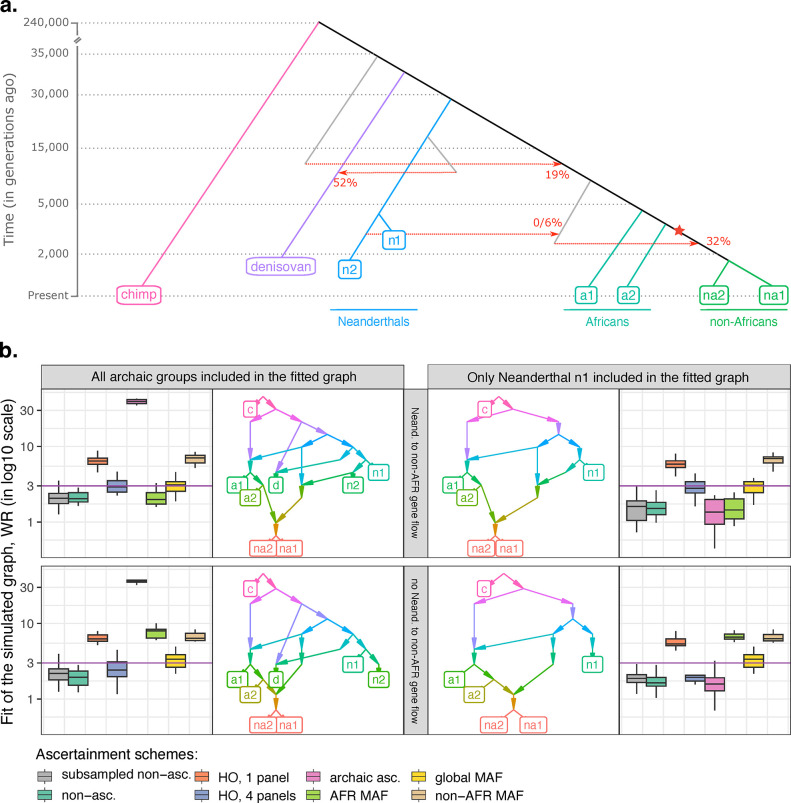
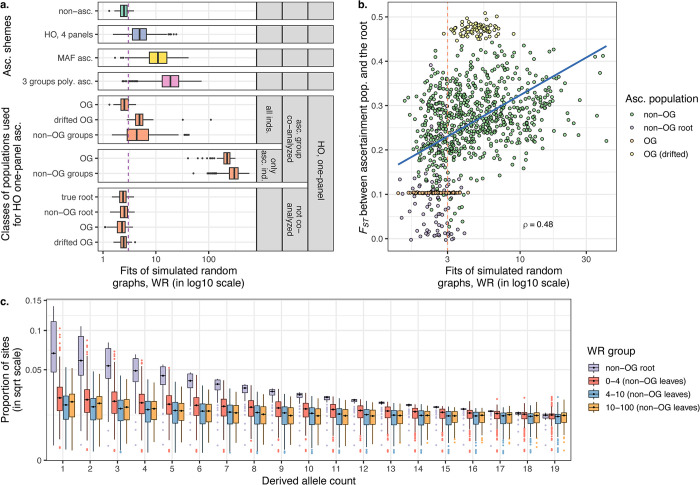
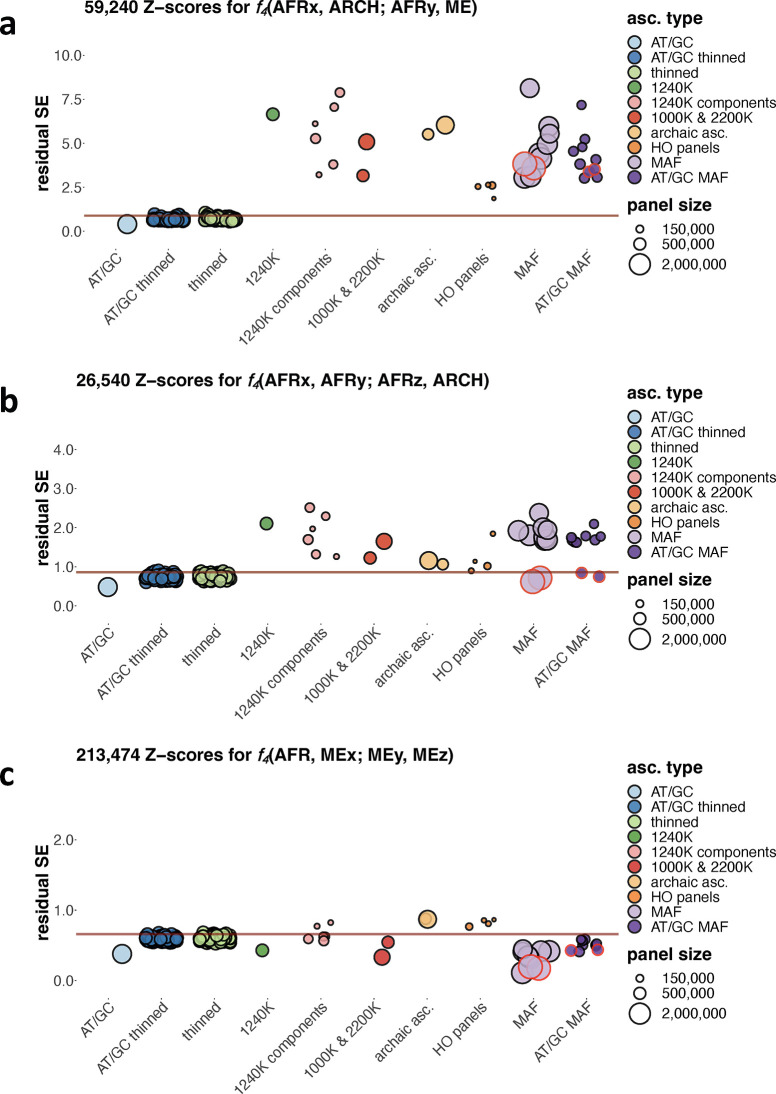
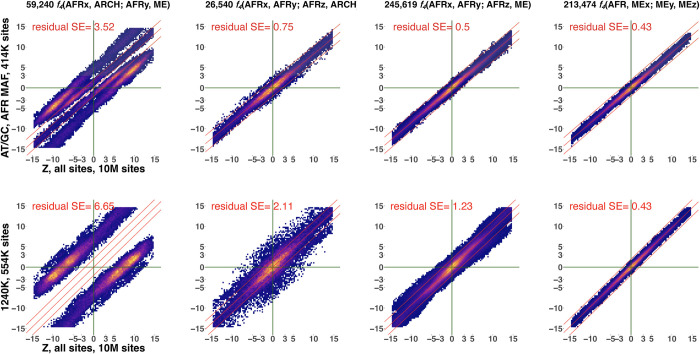
f-statistics have emerged as a first line of analysis for making inferences about demographic history from genome-wide data. Not only are they guaranteed to allow robust tests of the fits of proposed models of population history to data when analyzing full genome sequencing data-that is, all single nucleotide polymorphisms (SNPs) in the individuals being analyzed-but they are also guaranteed to allow robust tests of models for SNPs ascertained as polymorphic in a population that is an outgroup in a phylogenetic sense to all groups being analyzed. True "outgroup ascertainment" is in practice impossible in humans because our species has arisen from a substructured ancestral population that does not descend from a homogeneous ancestral population going back many hundreds of thousands of years into the past. However, initial studies suggested that non-outgroup-ascertainment schemes might produce robust enough results using f-statistics, and that motivated widespread fitting of models to data using non-outgroup-ascertained SNP panels such as the "Affymetrix Human Origins array" which has been genotyped on thousands of modern individuals from hundreds of populations, or the "1240k" in-solution enrichment reagent which has been the source of about 70% of published genome-wide data for ancient humans. In this study, we show that while analyses of population history using such panels work well for studies of relationships among non-African populations and one African outgroup, when co-modeling more than one sub-Saharan African and/or archaic human groups (Neanderthals and Denisovans), fitting of f-statistics to such SNP sets is expected to frequently lead to false rejection of true demographic histories, and failure to reject incorrect models. Analyzing panels of SNPs polymorphic in archaic humans, which has been suggested as a solution for the ascertainment problem, has limited statistical power and retains important biases. However, by carrying out simulations of diverse demographic histories, we show that bias in inferences based on f-statistics can be minimized by ascertaining on variants common in a union of diverse African groups; such ascertainment retains high statistical power while allowing co-analysis of archaic and modern groups.
Copyright: © 2023 Flegontov et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Modeling of African population history using f -statistics can be highly biased and is not addressed by previously suggested SNP ascertainment schemes.bioRxiv [Preprint]. 2023 Jan 22:2023.01.22.525077. doi: 10.1101/2023.01.22.525077. bioRxiv. 2023. Update in: PLoS Genet. 2023 Sep 7;19(9):e1010931. doi: 10.1371/journal.pgen.1010931. PMID: 36711923 Free PMC article. Updated. Preprint.
Similar articles
-
Modeling of African population history using f -statistics can be highly biased and is not addressed by previously suggested SNP ascertainment schemes.bioRxiv [Preprint]. 2023 Jan 22:2023.01.22.525077. doi: 10.1101/2023.01.22.525077. bioRxiv. 2023. Update in: PLoS Genet. 2023 Sep 7;19(9):e1010931. doi: 10.1371/journal.pgen.1010931. PMID: 36711923 Free PMC article. Updated. Preprint.
-
How do SNP ascertainment schemes and population demographics affect inferences about population history?BMC Genomics. 2015 Apr 3;16(1):266. doi: 10.1186/s12864-015-1469-5. BMC Genomics. 2015. PMID: 25887858 Free PMC article.
-
Neanderthal and Denisova genetic affinities with contemporary humans: introgression versus common ancestral polymorphisms.Gene. 2013 Nov 1;530(1):83-94. doi: 10.1016/j.gene.2013.06.005. Epub 2013 Jul 19. Gene. 2013. PMID: 23872234
-
Archaic human genomics.Am J Phys Anthropol. 2012;149 Suppl 55:24-39. doi: 10.1002/ajpa.22159. Epub 2012 Nov 2. Am J Phys Anthropol. 2012. PMID: 23124308 Review.
-
SNP ascertainment bias in population genetic analyses: why it is important, and how to correct it.Bioessays. 2013 Sep;35(9):780-6. doi: 10.1002/bies.201300014. Epub 2013 Jul 9. Bioessays. 2013. PMID: 23836388 Free PMC article. Review.
Cited by
-
Genomic exploration of the journey of Plasmodium vivax in Latin America.PLoS Pathog. 2025 Jan 13;21(1):e1012811. doi: 10.1371/journal.ppat.1012811. eCollection 2025 Jan. PLoS Pathog. 2025. PMID: 39804931 Free PMC article.
-
Performance of qpAdm-based screens for genetic admixture on graph-shaped histories and stepping stone landscapes.Genetics. 2025 May 8;230(1):iyaf047. doi: 10.1093/genetics/iyaf047. Genetics. 2025. PMID: 40169722 Free PMC article.
-
An explanation for the sister repulsion phenomenon in Patterson's f-statistics.Genetics. 2024 Nov 6;228(3):iyae144. doi: 10.1093/genetics/iyae144. Genetics. 2024. PMID: 39292210 Free PMC article.
-
The genomic footprints of migration: how ancient DNA reveals our history of mobility.Genome Biol. 2025 Jul 16;26(1):206. doi: 10.1186/s13059-025-03664-w. Genome Biol. 2025. PMID: 40671036 Free PMC article. Review.
-
Performance of qpAdm-based screens for genetic admixture on admixture-graph-shaped histories and stepping-stone landscapes.bioRxiv [Preprint]. 2025 Feb 3:2023.04.25.538339. doi: 10.1101/2023.04.25.538339. bioRxiv. 2025. Update in: Genetics. 2025 May 8;230(1):iyaf047. doi: 10.1093/genetics/iyaf047. PMID: 37904998 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
