

Podocyte Injury in Diabetic Kidney Disease in Mouse Models Involves TRPC6-mediated Calpain Activation Impairing Autophagy
- PMID: 37678257
- PMCID: PMC10631601
- DOI: 10.1681/ASN.0000000000000212
Podocyte Injury in Diabetic Kidney Disease in Mouse Models Involves TRPC6-mediated Calpain Activation Impairing Autophagy
Abstract
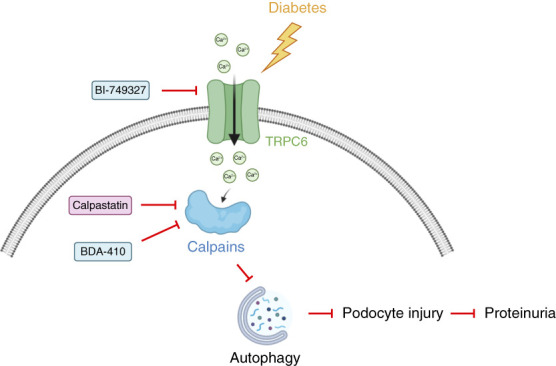
Significance statement: Autophagy protects podocytes from injury in diabetic kidney disease (DKD). Restoring glomerular autophagy is a promising approach to limit DKD. This study demonstrates a novel regulatory mechanism of autophagy that blocks this critical protection of the glomerular filtration barrier. We demonstrated that TRPC6 induced in podocytes in mouse models of diabetes mediates calpain activation, thereby impairing podocyte autophagy, causing injury and accelerating DKD. Furthermore, this study provides proof of principle for druggable targets for DKD because restoration of podocyte autophagy by calpain inhibitors effectively limits glomerular destruction.
Background: Diabetic kidney disease is associated with impaired podocyte autophagy and subsequent podocyte injury. The regulation of podocyte autophagy is unique because it minimally uses the mTOR and AMPK pathways. Thus, the molecular mechanisms underlying the impaired autophagy in podocytes in diabetic kidney disease remain largely elusive.
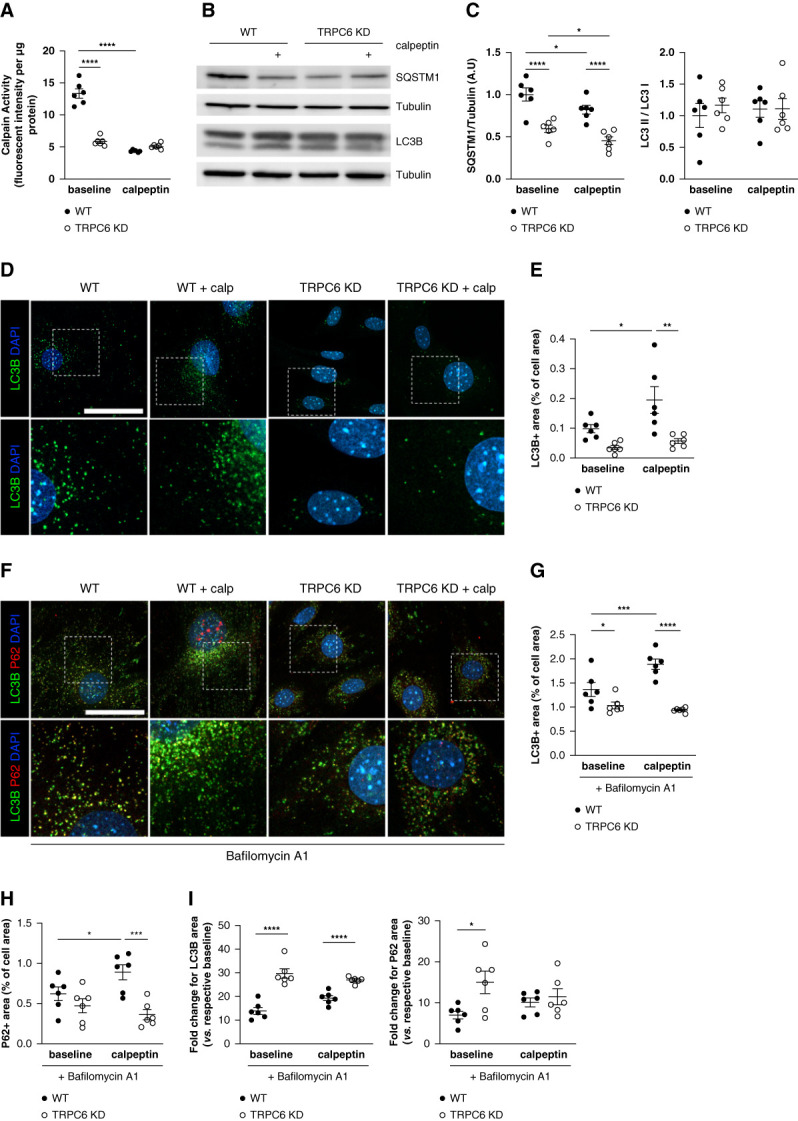
Methods: This study investigated how the calcium channel TRPC6 and the cysteine protease calpains deleteriously affect podocyte autophagy in diabetic kidney disease in mice. We demonstrated that TRPC6 knockdown in podocytes increased the autophagic flux because of decreased cysteine protease calpain activity. Diabetic kidney disease was induced in vivo using streptozotocin with unilateral nephrectomy and the BTBR ob/ob mouse models.
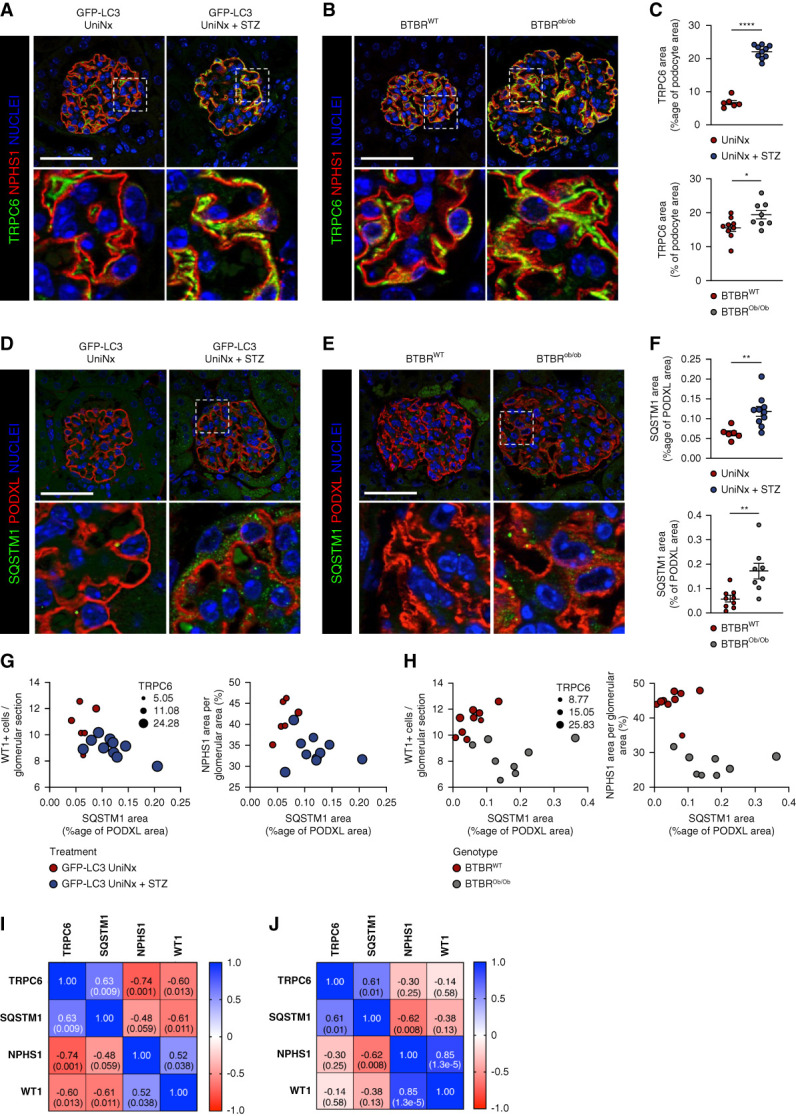
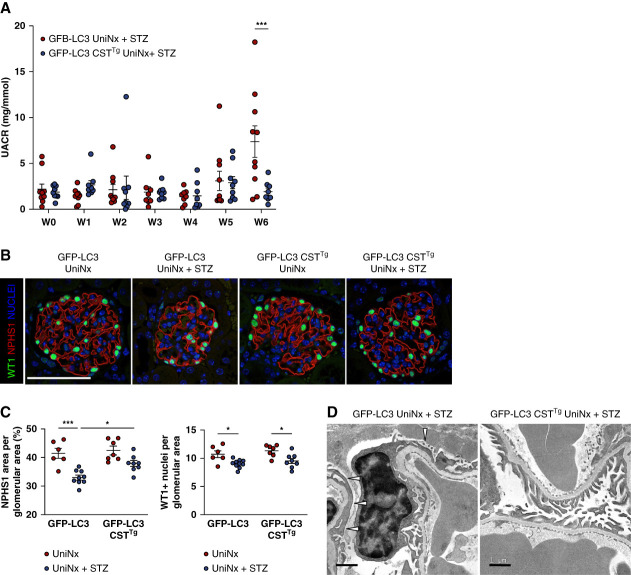
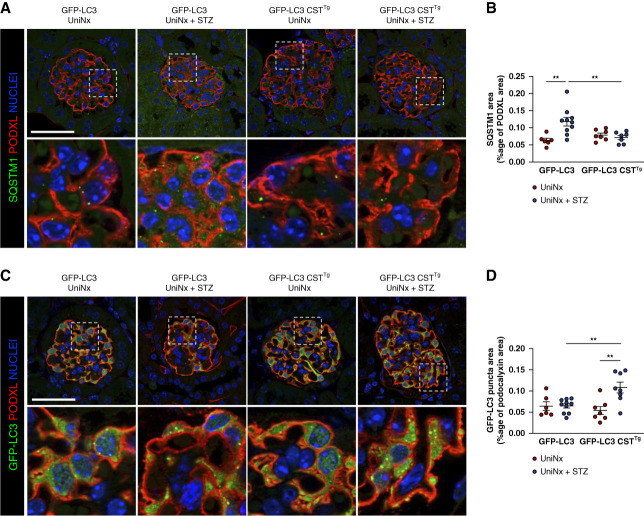
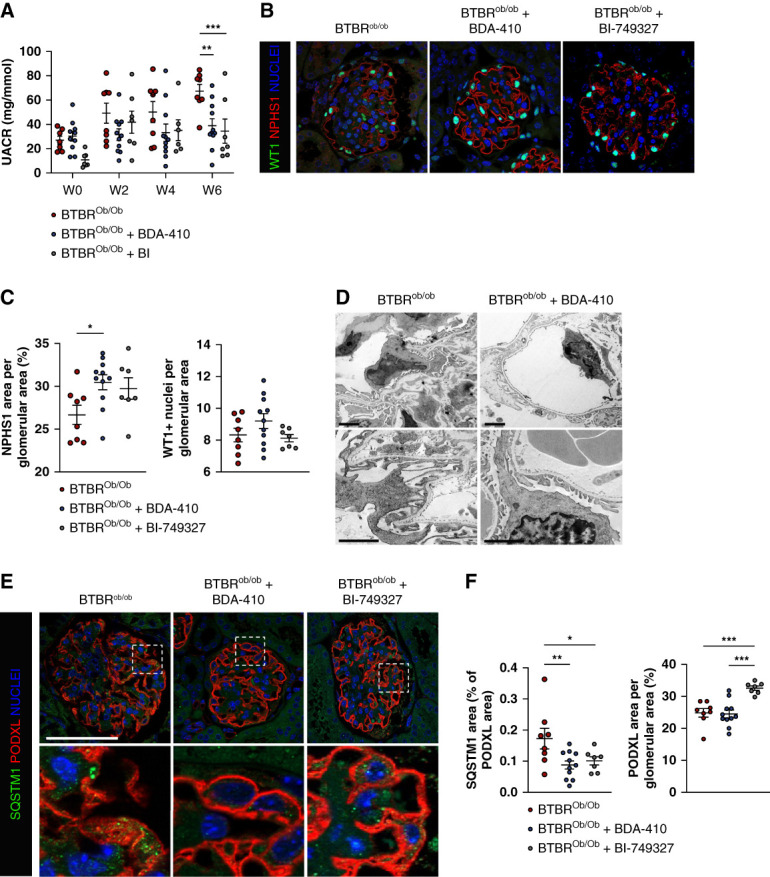
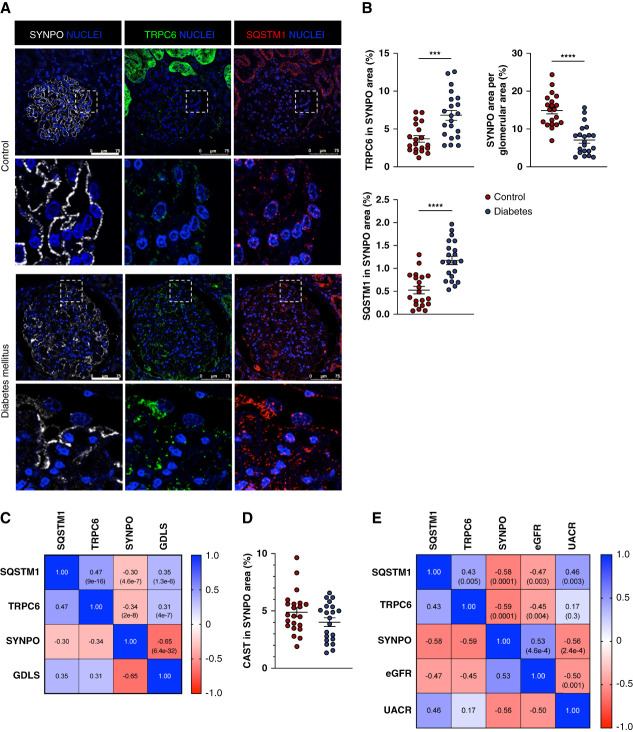
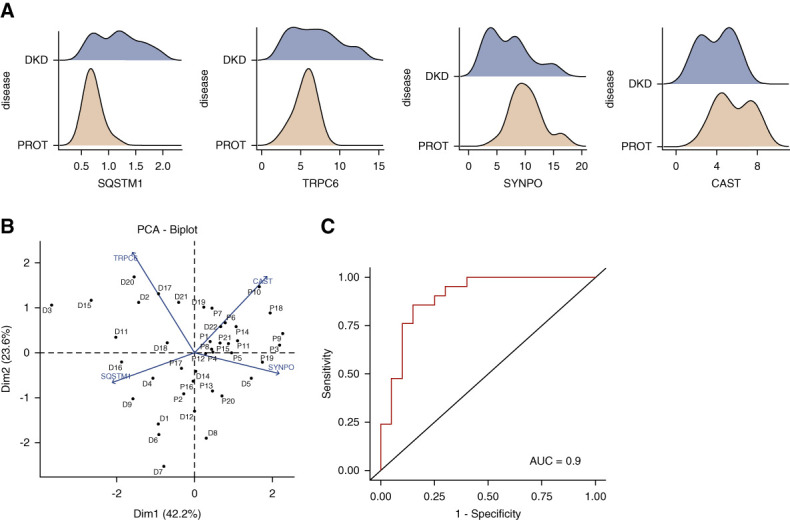
Results: Diabetes increased TRPC6 expression in podocytes in vivo with decreased podocyte autophagic flux. Transgenic overexpression of the endogenous calpain inhibitor calpastatin, as well as pharmacologic inhibition of calpain activity, normalized podocyte autophagic flux, reduced nephrin loss, and prevented the development of albuminuria in diabetic mice. In kidney biopsies from patients with diabetes, we further confirmed that TRPC6 overexpression in podocytes correlates with decreased calpastatin expression, autophagy blockade, and podocyte injury.
Conclusions: Overall, we discovered a new mechanism that connects TRPC6 and calpain activity to impaired podocyte autophagy, increased podocyte injury, and development of proteinuria in the context of diabetic kidney disease. Therefore, targeting TRPC6 and/or calpain to restore podocyte autophagy might be a promising therapeutic strategy for diabetic kidney disease.
Trial registration: ClinicalTrials.gov NCT05213624.
Copyright © 2023 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Society of Nephrology.
Conflict of interest statement
A. Karras reports Consultancy: Alnylam, GSK, Novartis, Otsuka, Vifor; Honoraria: ASTRAZENECA, Bohringer-Ingelheim, GSK, NOVARTIS, OTSUKA, PFIZER, VIFOR; Advisory or Leadership Role: Novartis, Otsuka, Vifor; and Speakers Bureau: AstraZeneca, Boehringer-Ingelheim, Otsuka, Pfizer, Vifor. E. Letavernier reports Consultancy: Biocodez, IKI, Withings; Research Funding: Advicenne, Coloplast; Honoraria: Biocodex, IKI, Withings; and Patents or Royalties: Insert transfert. T. Nijenhuis reports Research Funding: Dutch Kidney Foundation, Radboudumc; and Advisory or Leadership Role: Scientific board Dutch Society for Nephrology, Scientific advisory board Dutch Kidney Foundation, Speaker and chair Tubulopathies Expert Working Group and Executive Committee member, European Rare Kidney Disorders Network (ERKNet), and Board member ERA Working Group Genes & Kidney. M. Rabant reports Honoraria: Pfizer. R. Saito reports Employer: Mitsubishi Tanabe Pharma Corporation; and Other Interests or Relationships: IQ consortium; Life intelligence consortium (LINC). D.C. 't Hart reports Employer: Genome Diagnostics; Radboud university medical center. P.-L. Tharaux reports Consultancy: Alentis Therapeutics; Research Funding: Alentis Therapeutics; Honoraria: Travere Therapeutics; and Advisory or Leadership Role: French National Institute for Medical Research (INSERM), French Society of Cardiology (SFC), French Society of Hypertension (SFHTA), Associate Editor:
Figures
References
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
