

Microbial signature of plaque and gut in acute coronary syndrome
- PMID: 37679428
- PMCID: PMC10484905
- DOI: 10.1038/s41598-023-41867-y
Microbial signature of plaque and gut in acute coronary syndrome
Abstract
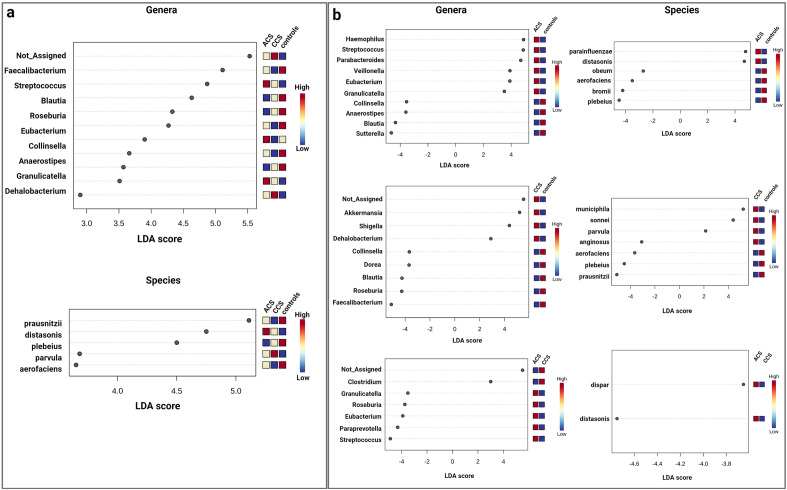
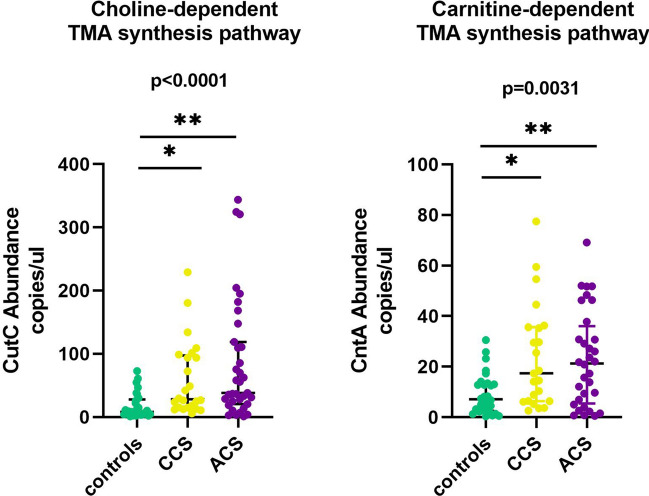
Gut microbiota is an emerging editable cardiovascular risk factor. We aim to investigate gut and coronary plaque microbiota, using fecal samples and angioplasty balloons from patients with acute coronary syndrome (ACS), chronic coronary syndrome (CCS) and control subjects. We examined bacterial communities in gut and coronary plaques by 16S rRNA sequencing and we performed droplet digital PCR analysis to investigate the gut relative abundance of the bacterial genes CutC/CntA involved in trimethylamine N-oxide synthesis. Linear discriminant analysis effect size (LEfSe) at the genus and species levels displayed gut enrichment in Streptococcus, Granulicatella and P. distasonis in ACS compared with CCS and controls; Roseburia, C. aerofaciens and F. prausnitzii were more abundant in controls than in patients. Principal component analysis (PCA) of 41 differentially abundant gut taxa showed a clustering of the three groups. In coronary plaque, LEfSe at the genus level revealed an enrichment of Staphylococcus and Streptococcus in ACS, and Paracoccus in CCS, whereas PCA of 15 differentially abundant plaque taxa exhibited clustering of ACS and CCS patients. CutC and CntA genes were more abundant in ACS and CCS than in controls while no significant difference emerged between ACS and CCS. Our results indicate that ACS and CCS exhibit a different gut and plaque microbial signature, suggesting a possible role of these microbiotas in coronary plaque instability.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Ardissino D, Berzuini C, Merlini PA, et al. Influence of 9p21.3 genetic variants on clinical and angiographic outcomes in early-onset myocardial infarction. J. Am. Coll. Cardiol. 2011;58:426–434. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
