

Design principles for cyclin K molecular glue degraders
- PMID: 37679459
- PMCID: PMC10746543
- DOI: 10.1038/s41589-023-01409-z
Design principles for cyclin K molecular glue degraders
Abstract
Molecular glue degraders are an effective therapeutic modality, but their design principles are not well understood. Recently, several unexpectedly diverse compounds were reported to deplete cyclin K by linking CDK12-cyclin K to the DDB1-CUL4-RBX1 E3 ligase. Here, to investigate how chemically dissimilar small molecules trigger cyclin K degradation, we evaluated 91 candidate degraders in structural, biophysical and cellular studies and reveal all compounds acquire glue activity via simultaneous CDK12 binding and engagement of DDB1 interfacial residues, in particular Arg928. While we identify multiple published kinase inhibitors as cryptic degraders, we also show that these glues do not require pronounced inhibitory properties for activity and that the relative degree of CDK12 inhibition versus cyclin K degradation is tuneable. We further demonstrate cyclin K degraders have transcriptional signatures distinct from CDK12 inhibitors, thereby offering unique therapeutic opportunities. The systematic structure-activity relationship analysis presented herein provides a conceptual framework for rational molecular glue design.
© 2023. The Author(s).
Conflict of interest statement
N.H.T. receives funding from the Novartis Research Foundation and is a scientific advisory board (SAB) member of Monte Rosa Therapeutics and an advisor to Zenith Therapeutics and Ridgeline. B.L.E. has received research funding from Celgene, Deerfield, Novartis and Calico. He has received consulting fees from GRAIL, and he serves on the SABs for and holds equity in Neomorph, TenSixteen Bio, Skyhawk Therapeutics and Exo Therapeutics. E.S.F. is a founder, SAB member and equity holder of Civetta Therapeutics, Lighthorse Therapeutics, Proximity Therapeutics and Neomorph, Inc. (board member). E.S.F. is an equity holder and SAB member for Avilar Therapeutics, Ajax Therapeutics and Photys Therapeutics and a consultant to Novartis, Sanofi, EcoR1 Capital, Odyssey, Astellas and Deerfield. The Fischer lab receives or has received research funding from Novartis, Ajax, Voronoi, Interline, Deerfield and Astellas. D.G. is an SAB member of FoRx therapeutics. The C.M.-R. lab receives research funding from Almirall and Aelin Tx, and C.M.-R. is an SAB member of Nostrum Biodiscovery. G.E.W. is scientific founder and shareholder of Proxygen and Solgate, the Winter laboratory receives research funding from Pfizer. M.S. has received research funding from Calico Life Sciences LLC. G.P. is now an employee of Monte Rosa Therapeutics. C.D.G. is now an employee of VantAI. The remaining authors declare no competing interests.
Figures
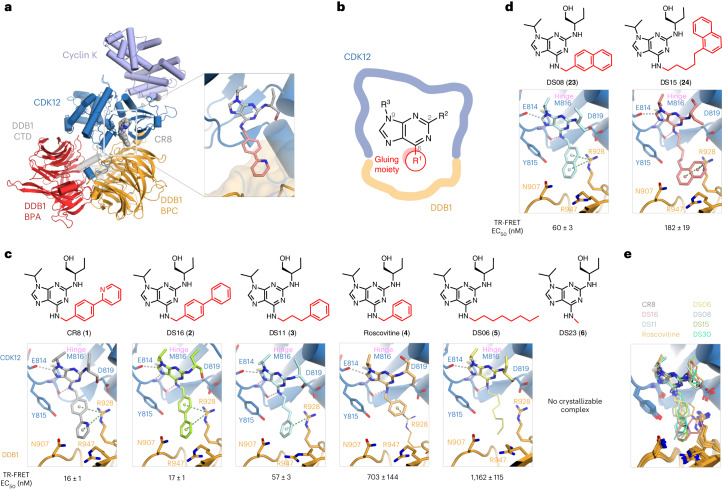

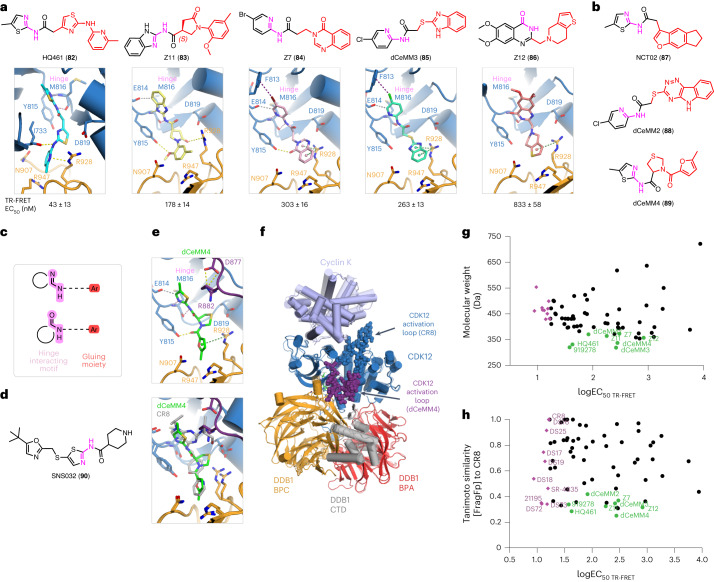
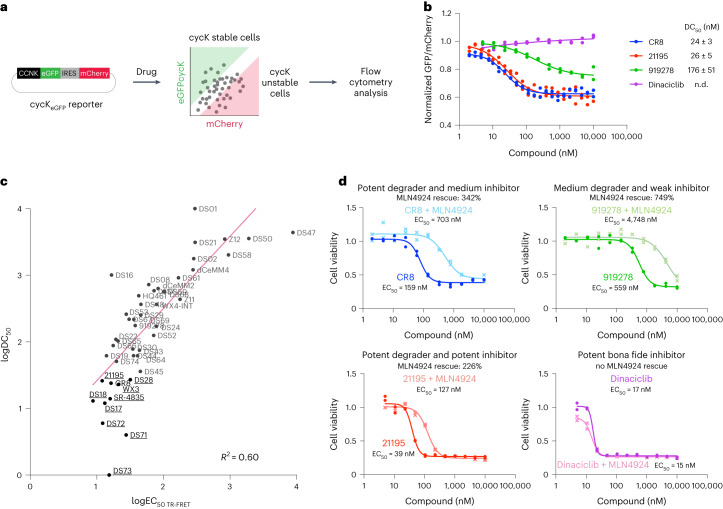
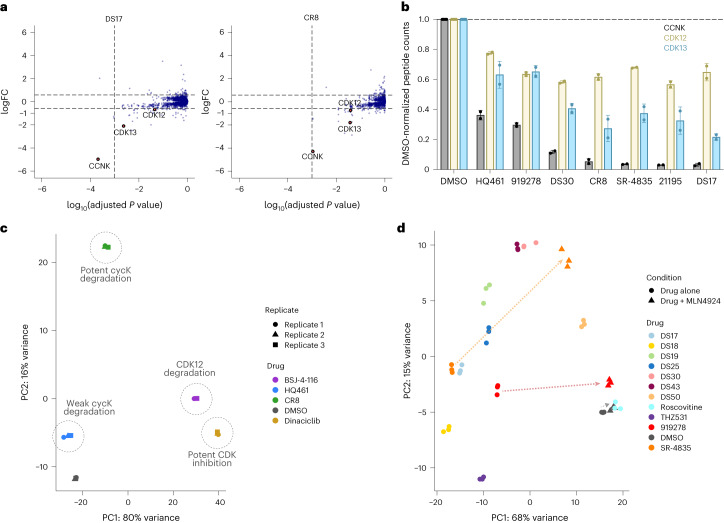
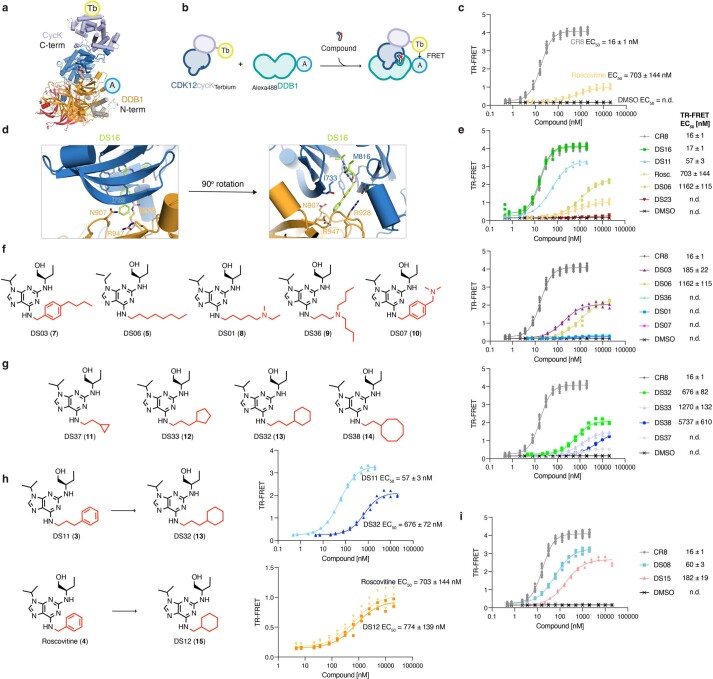




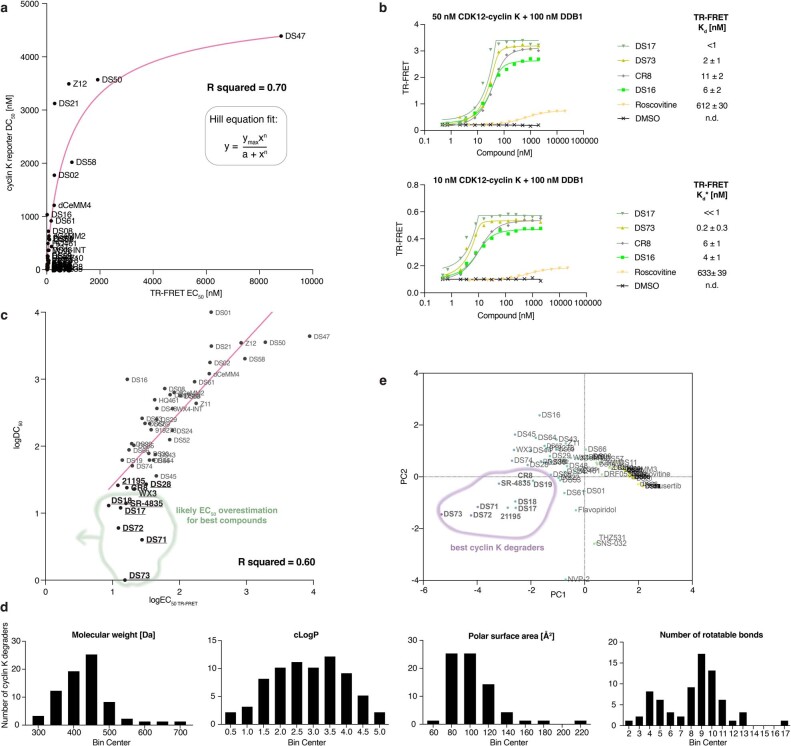

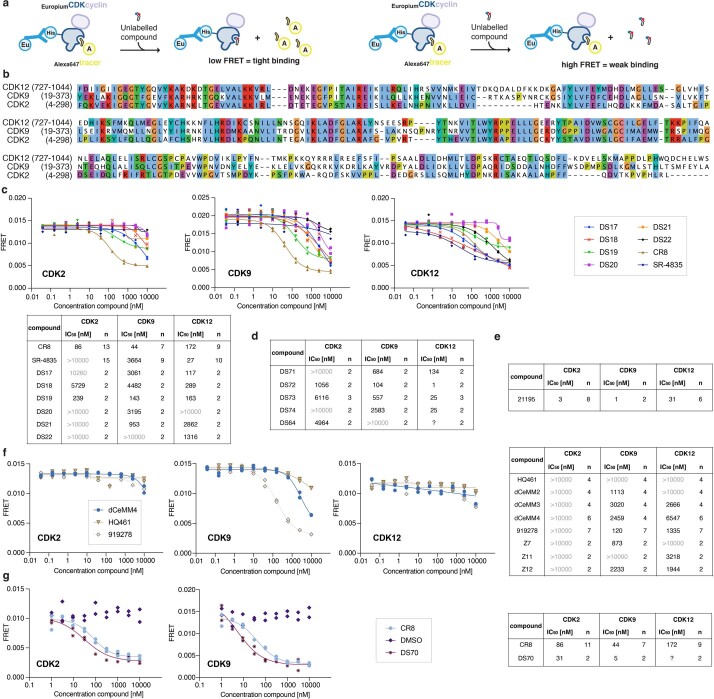

References
-
- Cowan, A. D. & Ciulli, A. Driving E3 ligase substrate specificity for targeted protein degradation: lessons from nature and the laboratory. Annu. Rev. Biochem. 10.1146/annurev-biochem-032620-104421 (2022). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
