

Multi-omics data integration using ratio-based quantitative profiling with Quartet reference materials
- PMID: 37679543
- PMCID: PMC11252085
- DOI: 10.1038/s41587-023-01934-1
Multi-omics data integration using ratio-based quantitative profiling with Quartet reference materials
Abstract
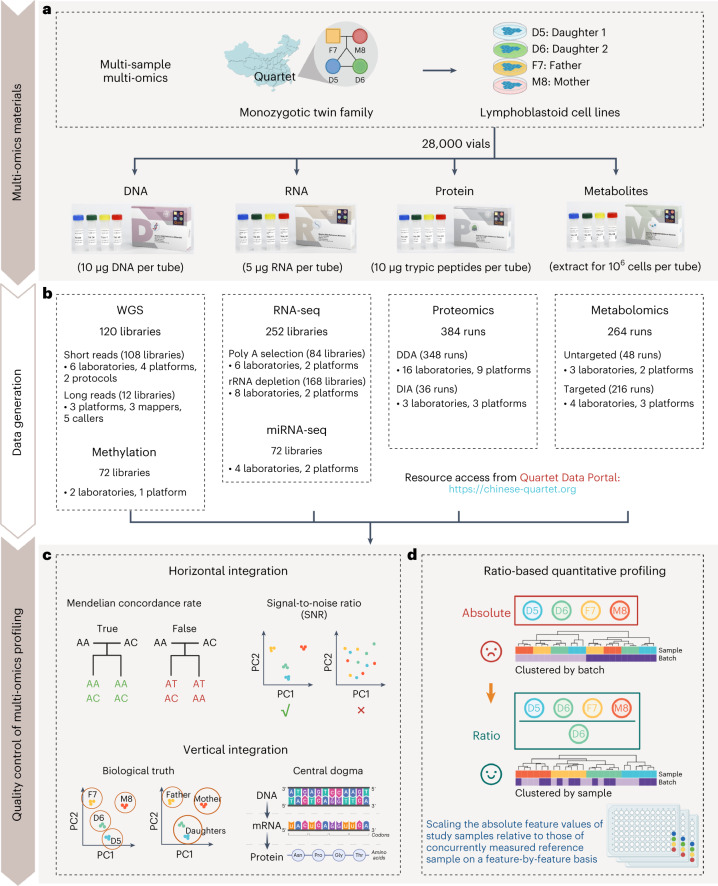
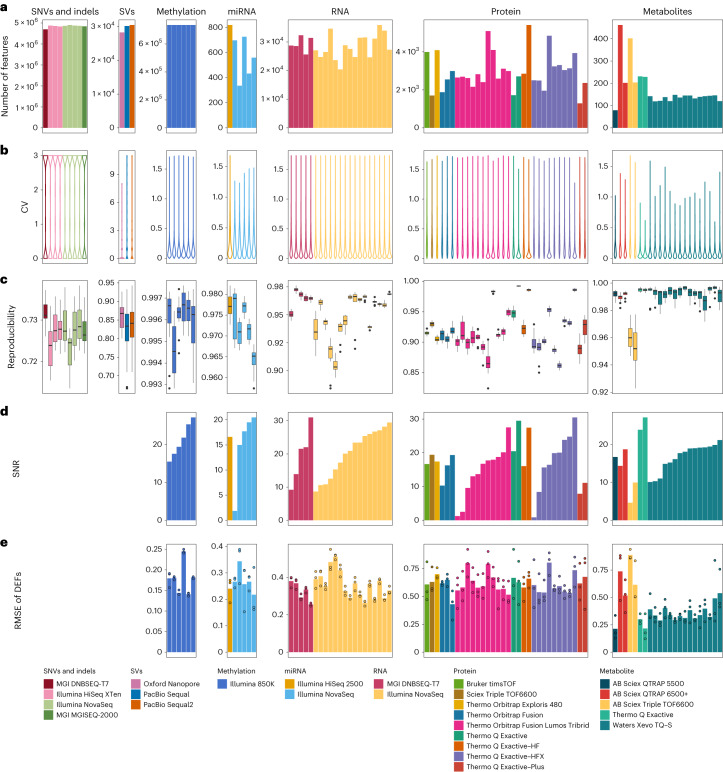
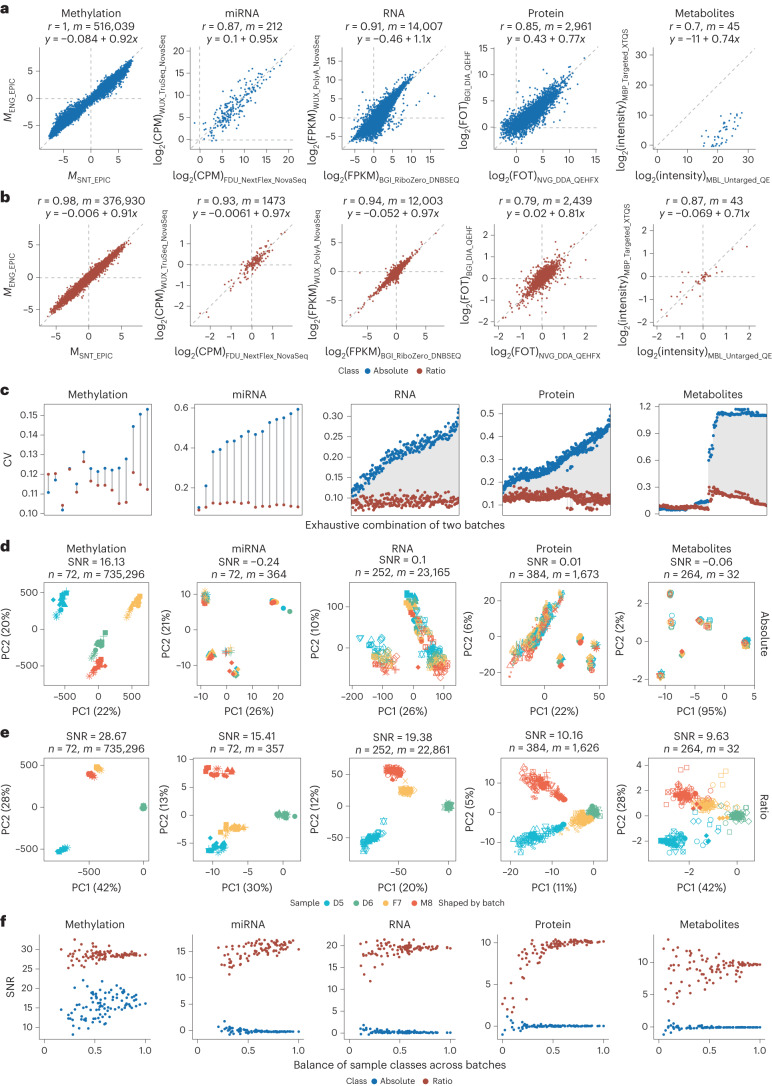
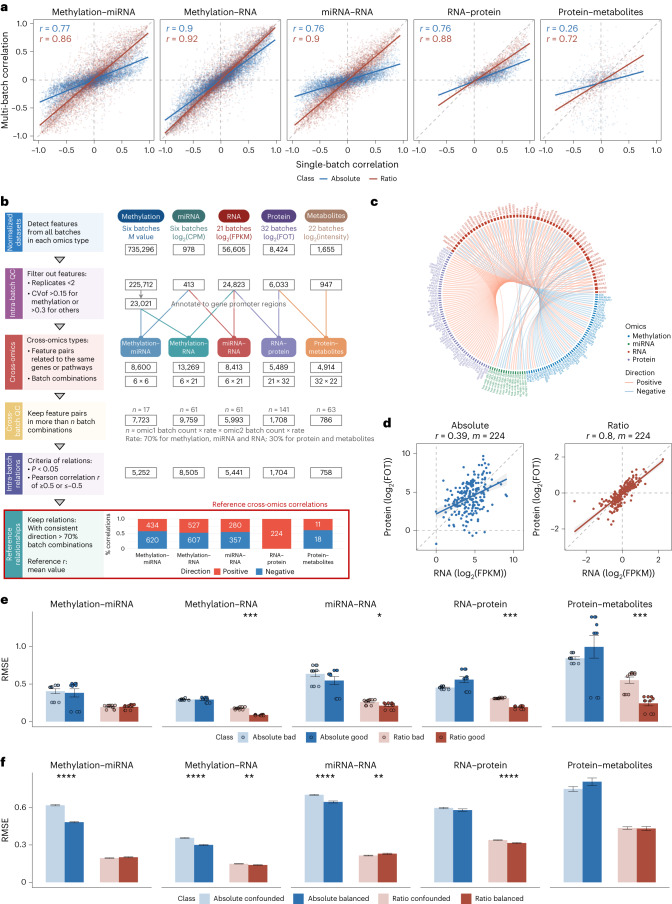
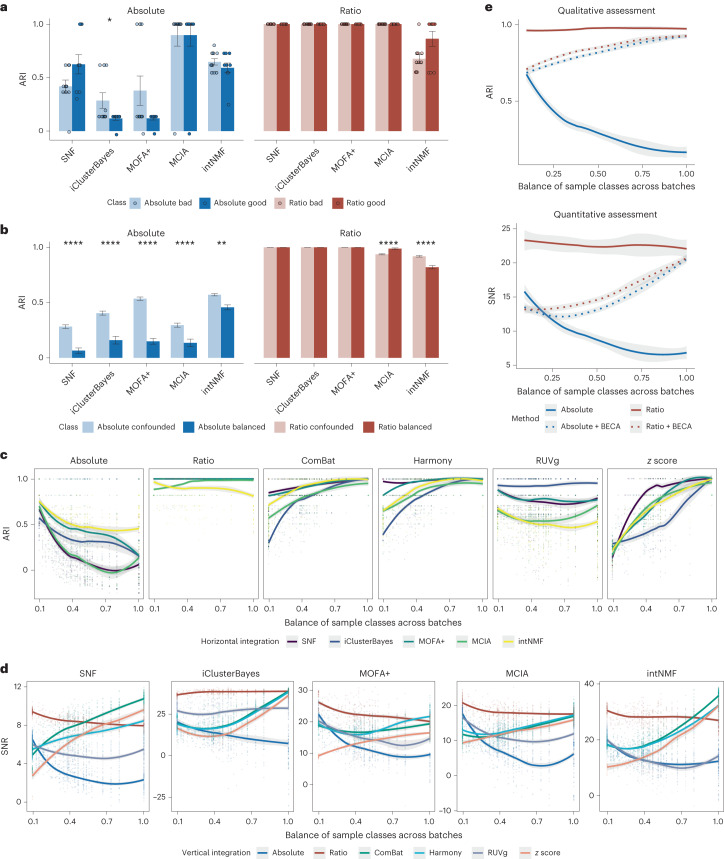
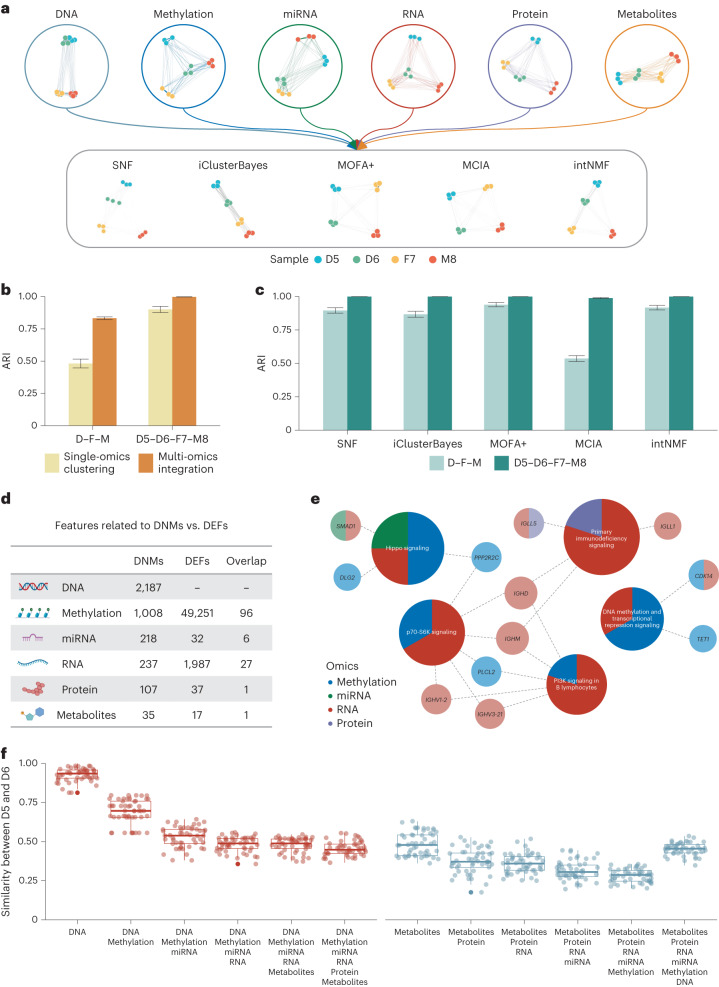
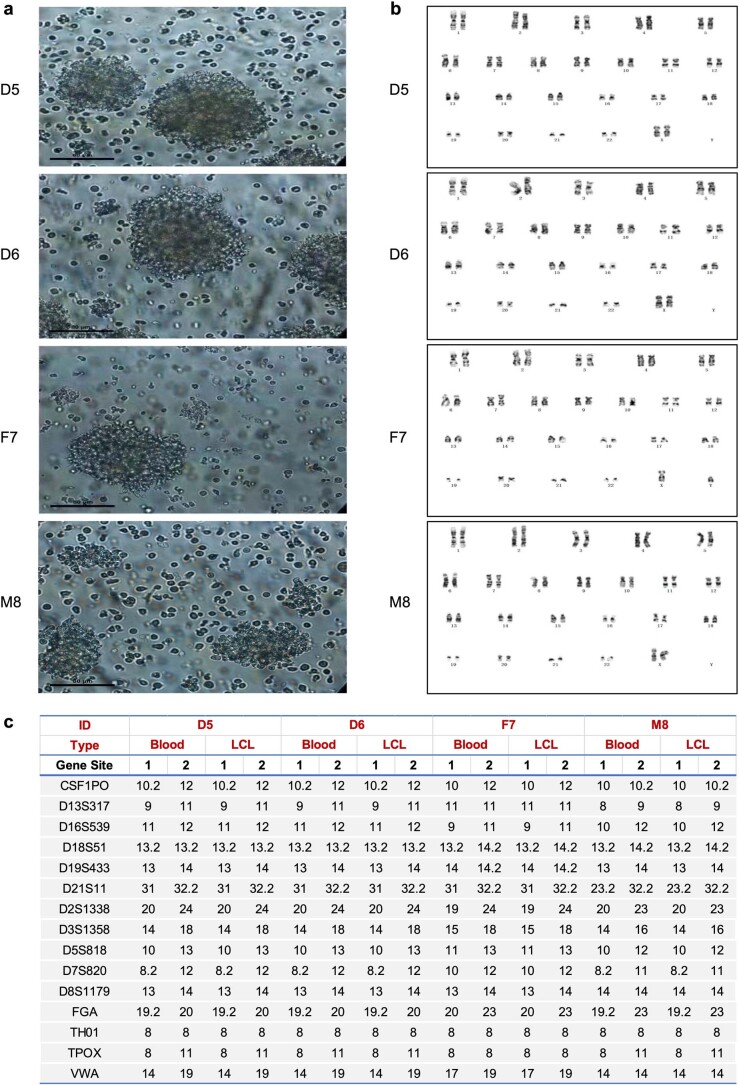
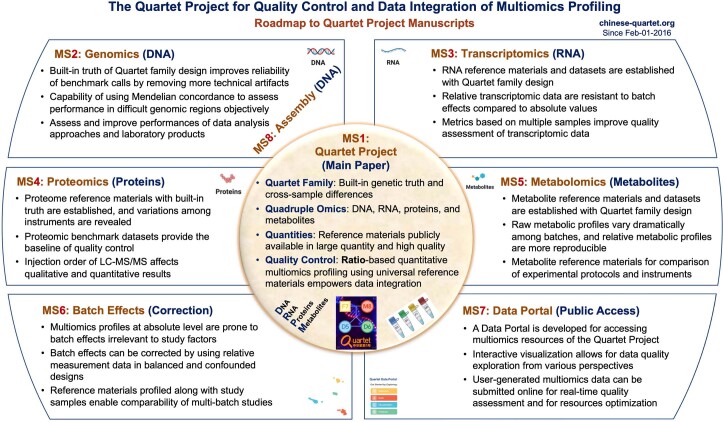
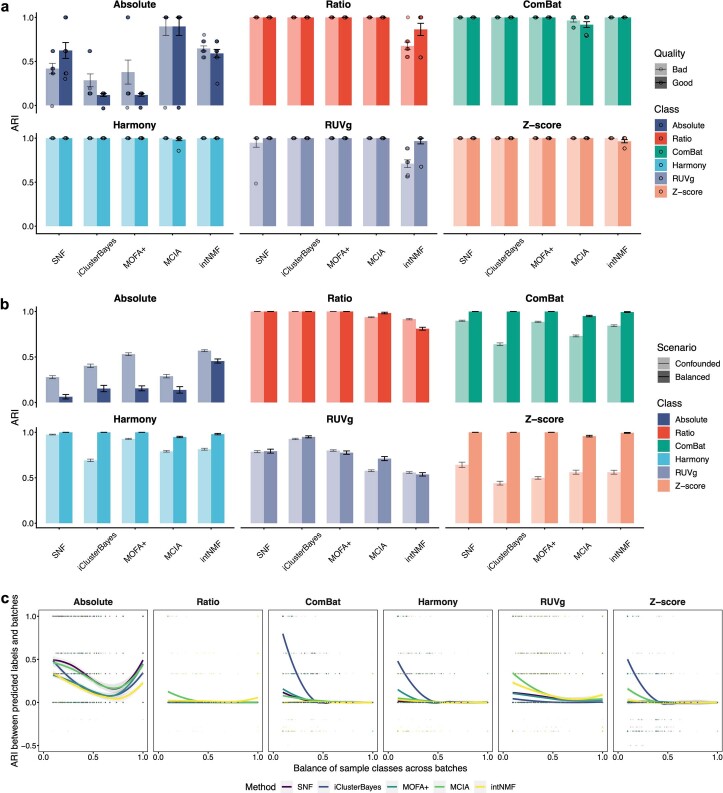
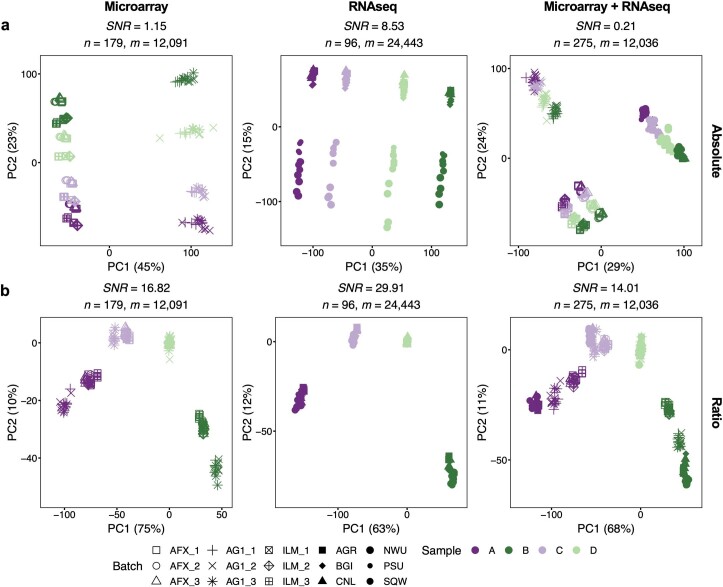
Characterization and integration of the genome, epigenome, transcriptome, proteome and metabolome of different datasets is difficult owing to a lack of ground truth. Here we develop and characterize suites of publicly available multi-omics reference materials of matched DNA, RNA, protein and metabolites derived from immortalized cell lines from a family quartet of parents and monozygotic twin daughters. These references provide built-in truth defined by relationships among the family members and the information flow from DNA to RNA to protein. We demonstrate how using a ratio-based profiling approach that scales the absolute feature values of a study sample relative to those of a concurrently measured common reference sample produces reproducible and comparable data suitable for integration across batches, labs, platforms and omics types. Our study identifies reference-free 'absolute' feature quantification as the root cause of irreproducibility in multi-omics measurement and data integration and establishes the advantages of ratio-based multi-omics profiling with common reference materials.
© 2023. The Author(s).
Conflict of interest statement
J.H. is an employee of Vazyme Biotech Co. Ltd. L.Z. is the cofounder of Vazyme Biotech Co. Ltd. Hui Jiang is an employee of MGI, BGI-Shenzhen. All other authors declare no competing interests.
Figures
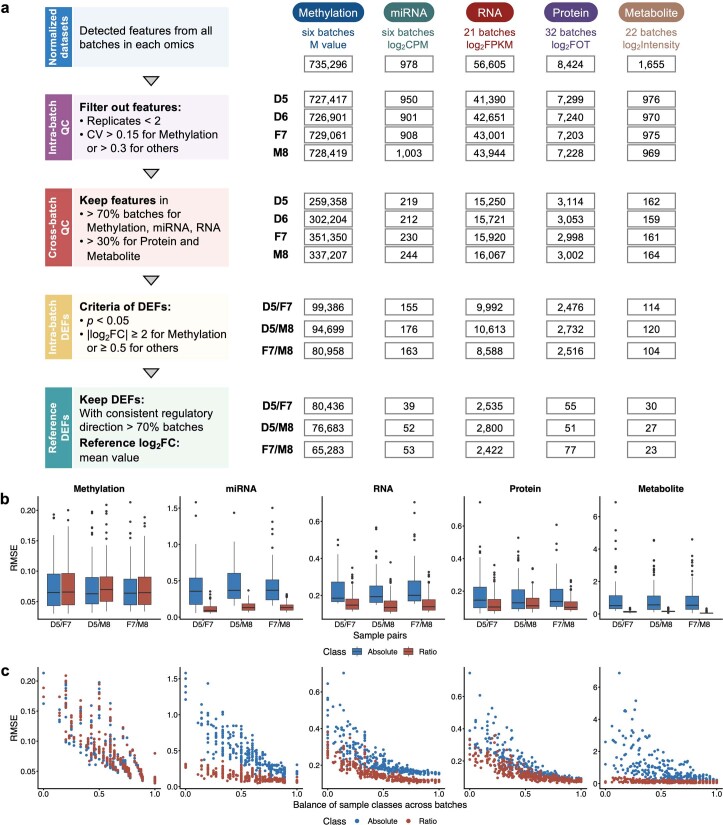
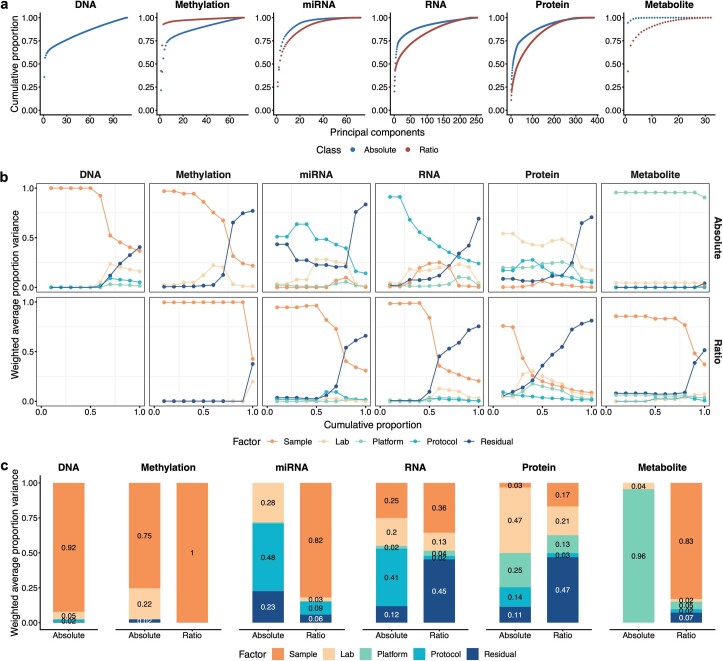
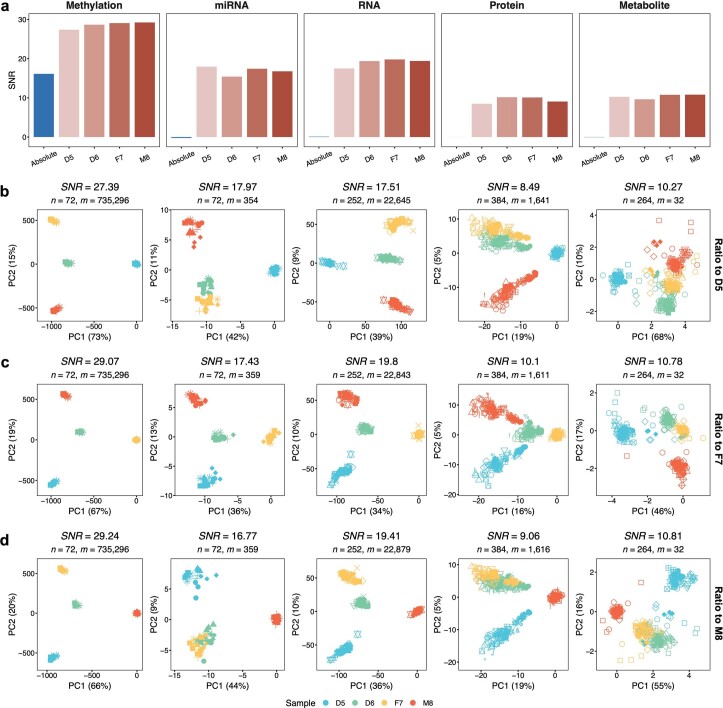
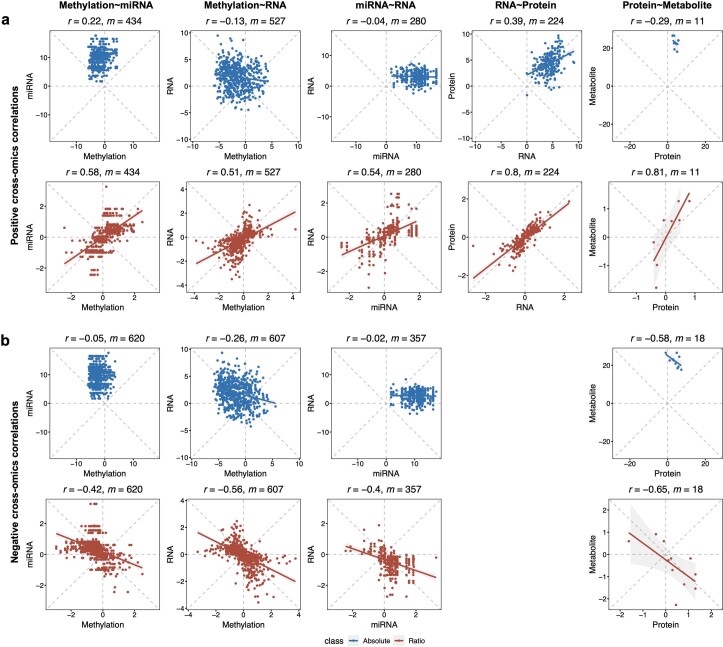
References
-
- Shilo, S., Rossman, H. & Segal, E. Axes of a revolution: challenges and promises of big data in healthcare. Nat. Med.26, 29–38 (2020). - PubMed
-
- Ideker, T., Galitski, T. & Hood, L. A new approach to decoding life: systems biology. Annu. Rev. Genom. Hum. Genet.2, 343–372 (2001). - PubMed
-
- Wang, B. et al. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods11, 333–337 (2014). - PubMed
MeSH terms
Grants and funding
- 31720103909/National Natural Science Foundation of China (National Science Foundation of China)
- 32170657/National Natural Science Foundation of China (National Science Foundation of China)
- SKLGE-2117/State Key Laboratory of Genetic Engineering (SKLGE)
- 2017SHZDZX01/Science and Technology Commission of Shanghai Municipality (Shanghai Municipal Science and Technology Commission)
LinkOut - more resources
Full Text Sources
