

Hepatic FASN deficiency differentially affects nonalcoholic fatty liver disease and diabetes in mouse obesity models
- PMID: 37681411
- PMCID: PMC10544238
- DOI: 10.1172/jci.insight.161282
Hepatic FASN deficiency differentially affects nonalcoholic fatty liver disease and diabetes in mouse obesity models
Abstract
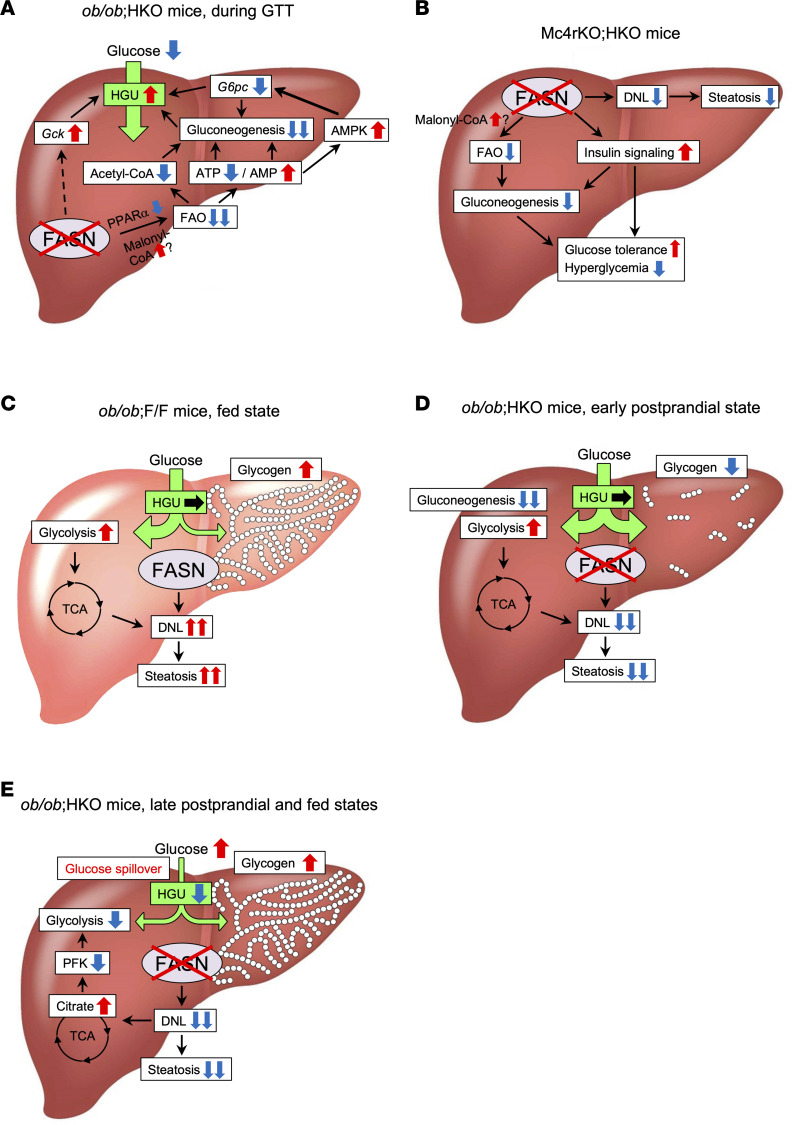
Nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes are interacting comorbidities of obesity, and increased hepatic de novo lipogenesis (DNL), driven by hyperinsulinemia and carbohydrate overload, contributes to their pathogenesis. Fatty acid synthase (FASN), a key enzyme of hepatic DNL, is upregulated in association with insulin resistance. However, the therapeutic potential of targeting FASN in hepatocytes for obesity-associated metabolic diseases is unknown. Here, we show that hepatic FASN deficiency differentially affects NAFLD and diabetes depending on the etiology of obesity. Hepatocyte-specific ablation of FASN ameliorated NAFLD and diabetes in melanocortin 4 receptor-deficient mice but not in mice with diet-induced obesity. In leptin-deficient mice, FASN ablation alleviated hepatic steatosis and improved glucose tolerance but exacerbated fed hyperglycemia and liver dysfunction. The beneficial effects of hepatic FASN deficiency on NAFLD and glucose metabolism were associated with suppression of DNL and attenuation of gluconeogenesis and fatty acid oxidation, respectively. The exacerbation of fed hyperglycemia by FASN ablation in leptin-deficient mice appeared attributable to impairment of hepatic glucose uptake triggered by glycogen accumulation and citrate-mediated inhibition of glycolysis. Further investigation of the therapeutic potential of hepatic FASN inhibition for NAFLD and diabetes in humans should thus consider the etiology of obesity.
Keywords: Diabetes; Hepatology; Metabolism; Molecular pathology; Obesity.
Figures







References
-
- Chalasani N, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55(6):2005–2023. doi: 10.1002/hep.25762. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
