

ATAC and SAGA co-activator complexes utilize co-translational assembly, but their cellular localization properties and functions are distinct
- PMID: 37682711
- PMCID: PMC10591836
- DOI: 10.1016/j.celrep.2023.113099
ATAC and SAGA co-activator complexes utilize co-translational assembly, but their cellular localization properties and functions are distinct
Abstract
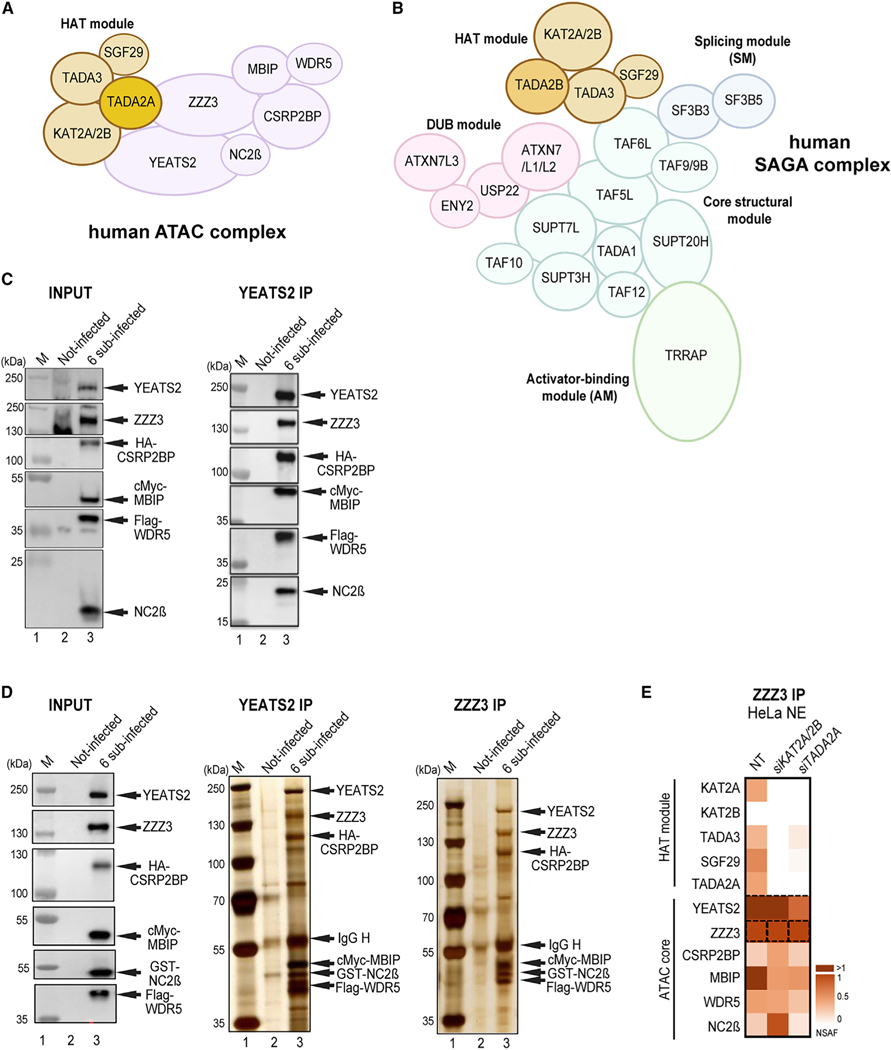
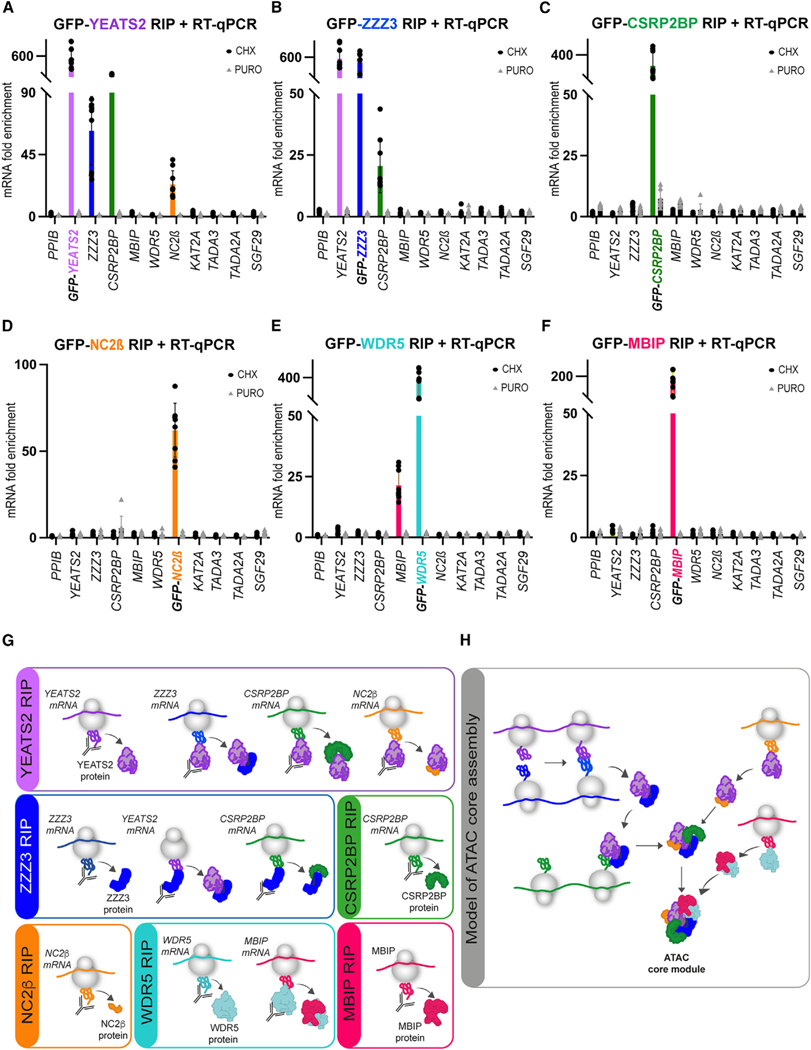
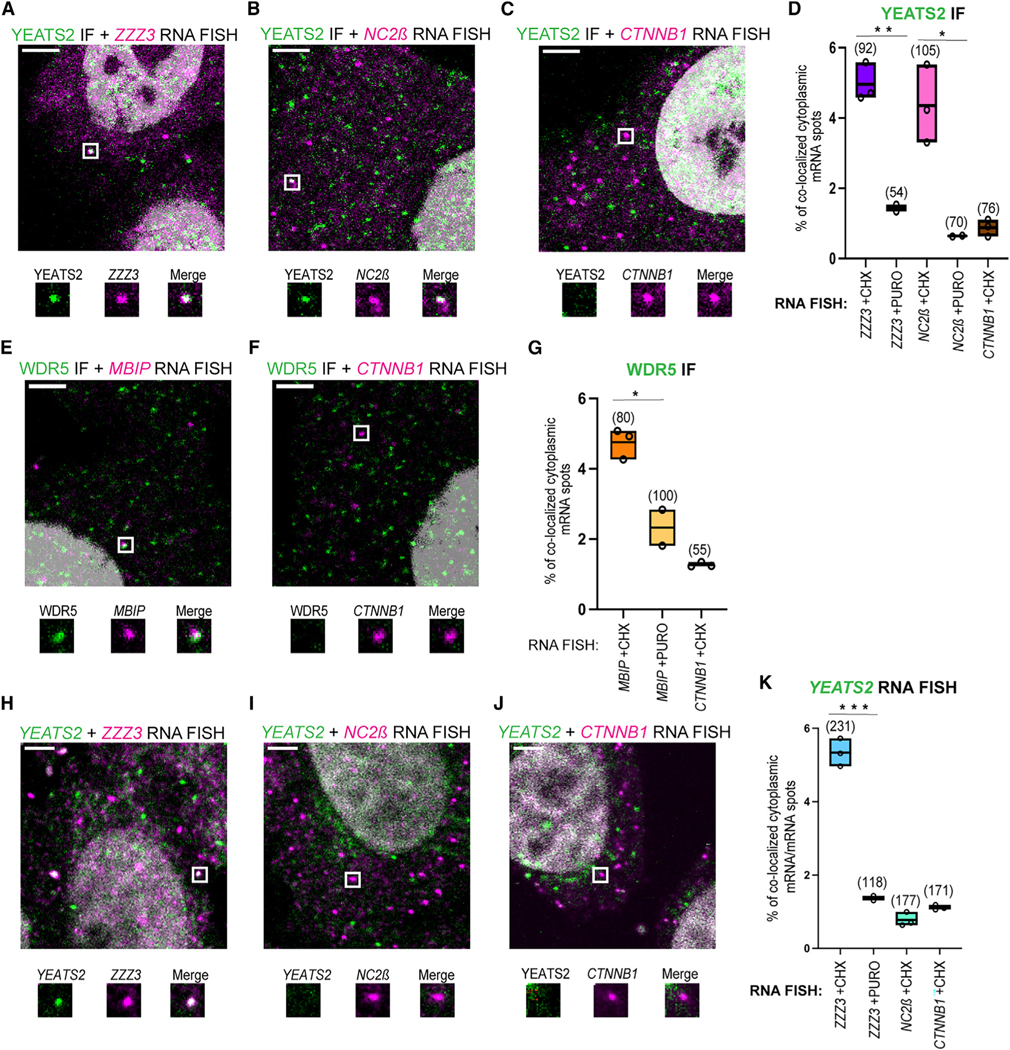
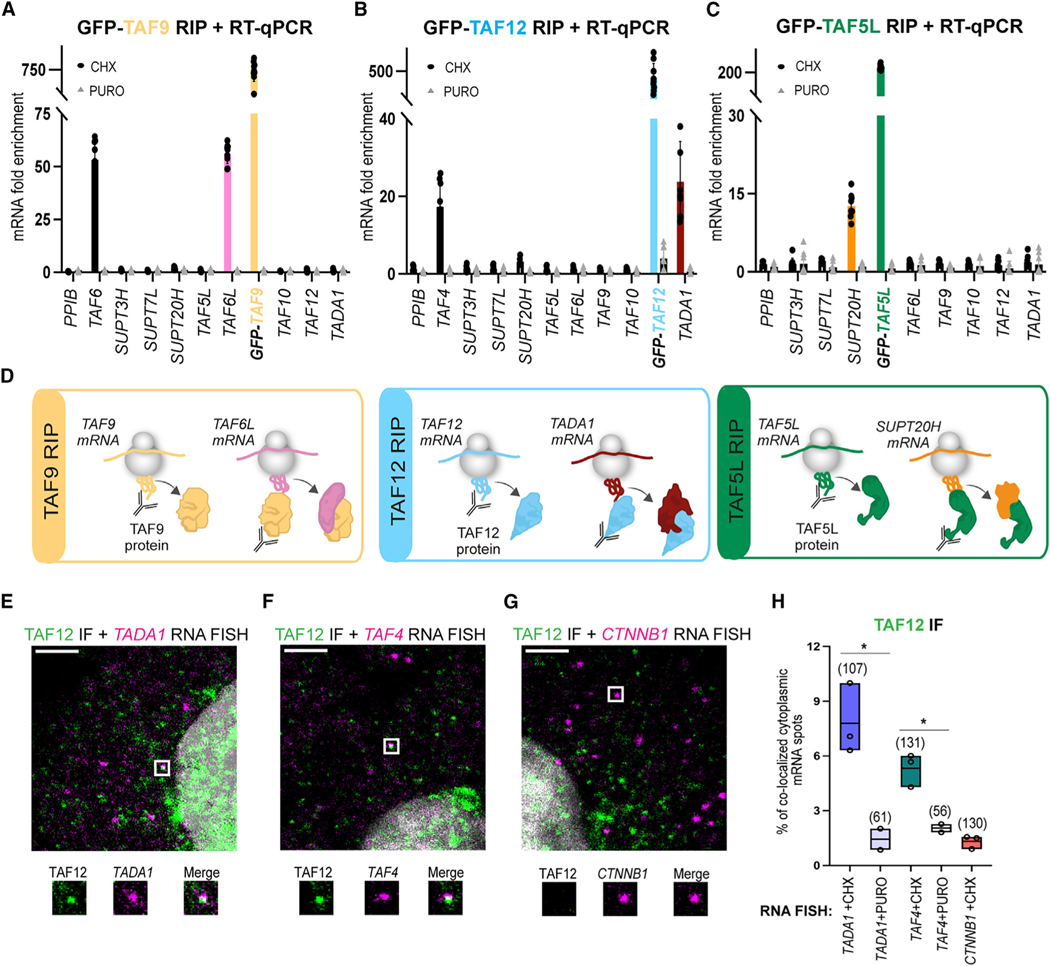
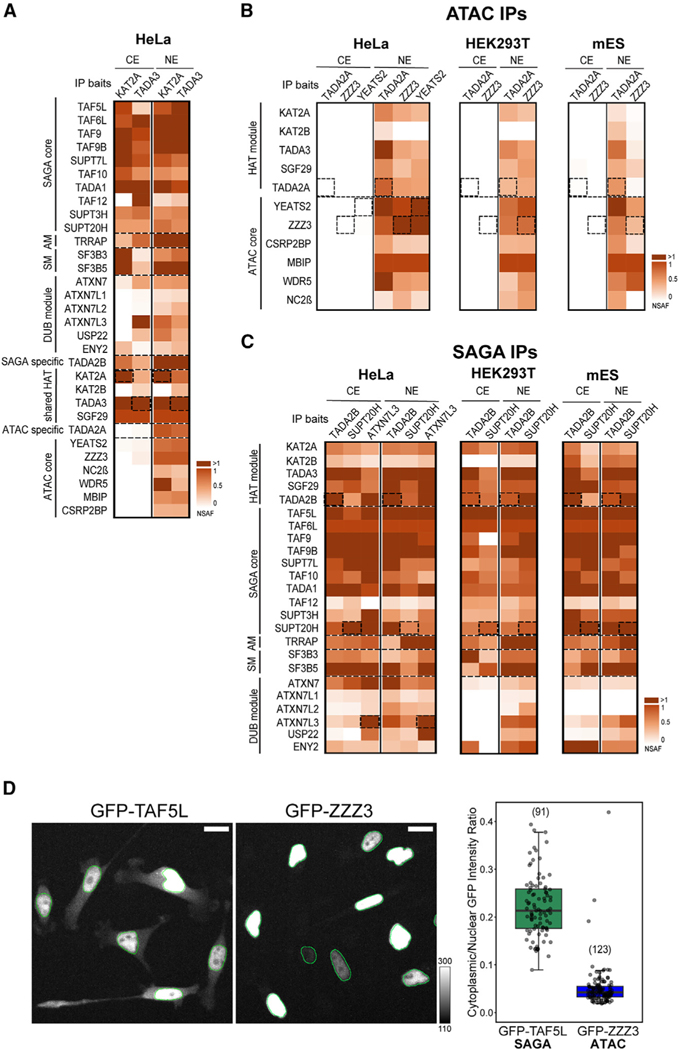
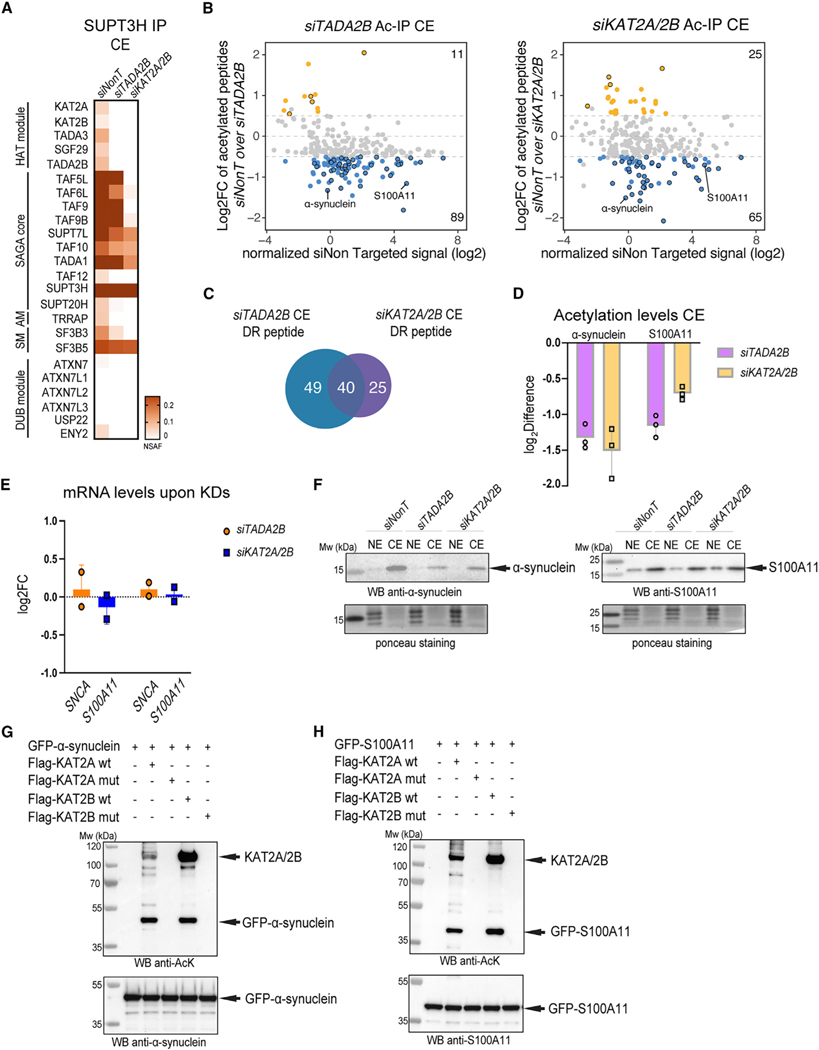
To understand the function of multisubunit complexes, it is of key importance to uncover the precise mechanisms that guide their assembly. Nascent proteins can find and bind their interaction partners during their translation, leading to co-translational assembly. Here, we demonstrate that the core modules of ATAC (ADA-two-A-containing) and SAGA (Spt-Ada-Gcn5-acetyltransferase), two lysine acetyl transferase-containing transcription co-activator complexes, assemble co-translationally in the cytoplasm of mammalian cells. In addition, a SAGA complex containing all of its modules forms in the cytoplasm and acetylates non-histone proteins. In contrast, ATAC complex subunits cannot be detected in the cytoplasm of mammalian cells. However, an endogenous ATAC complex containing two functional modules forms and functions in the nucleus. Thus, the two related co-activators, ATAC and SAGA, assemble using co-translational pathways, but their subcellular localization, cytoplasmic abundance, and functions are distinct.
Keywords: CP: Molecular biology; RIP; RNA immunoprecipitation; YEATS2; ZZZ3; biogenesis; co-translation; lysine acetylation; multiprotein complex; non-histone protein; ribosome; single-molecule RNA FISH; transcription regulation.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Update of
-
ATAC and SAGA coactivator complexes utilize co-translational assembly, but their cellular localization properties and functions are distinct.bioRxiv [Preprint]. 2023 Aug 3:2023.08.03.551787. doi: 10.1101/2023.08.03.551787. bioRxiv. 2023. Update in: Cell Rep. 2023 Sep 26;42(9):113099. doi: 10.1016/j.celrep.2023.113099. PMID: 37577620 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
