

Comparative genomics of the proteostasis network in extreme acidophiles
- PMID: 37682893
- PMCID: PMC10490939
- DOI: 10.1371/journal.pone.0291164
Comparative genomics of the proteostasis network in extreme acidophiles
Abstract
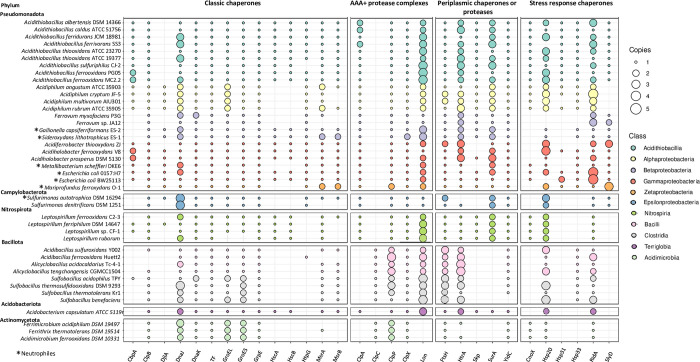
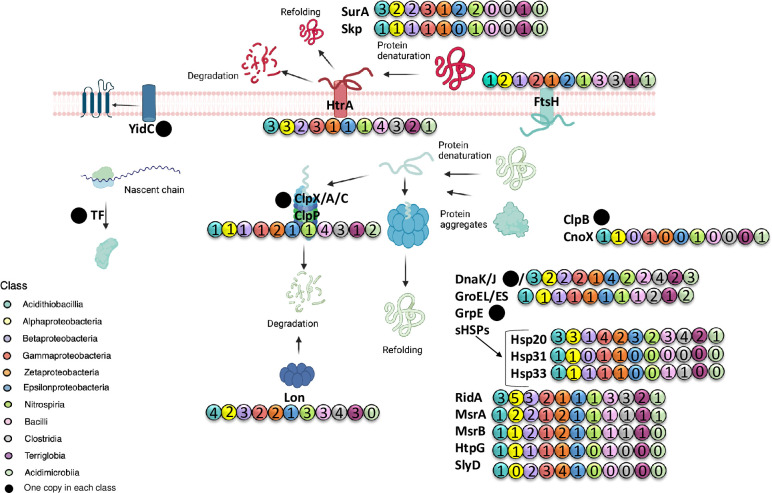
Extreme acidophiles thrive in harsh environments characterized by acidic pH, high concentrations of dissolved metals and high osmolarity. Most of these microorganisms are chemolithoautotrophs that obtain energy from low redox potential sources, such as the oxidation of ferrous ions. Under these conditions, the mechanisms that maintain homeostasis of proteins (proteostasis), as the main organic components of the cells, are of utmost importance. Thus, the analysis of protein chaperones is critical for understanding how these organisms deal with proteostasis under such environmental conditions. In this work, using a bioinformatics approach, we performed a comparative genomic analysis of the genes encoding classical, periplasmic and stress chaperones, and the protease systems. The analysis included 35 genomes from iron- or sulfur-oxidizing autotrophic, heterotrophic, and mixotrophic acidophilic bacteria. The results showed that classical ATP-dependent chaperones, mostly folding chaperones, are widely distributed, although they are sub-represented in some groups. Acidophilic bacteria showed redundancy of genes coding for the ATP-independent holdase chaperones RidA and Hsp20. In addition, a systematically high redundancy of genes encoding periplasmic chaperones like HtrA and YidC was also detected. In the same way, the proteolytic ATPase complexes ClpPX and Lon presented redundancy and broad distribution. The presence of genes that encoded protein variants was noticeable. In addition, genes for chaperones and protease systems were clustered within the genomes, suggesting common regulation of these activities. Finally, some genes were differentially distributed between bacteria as a function of the autotrophic or heterotrophic character of their metabolism. These results suggest that acidophiles possess an abundant and flexible proteostasis network that protects proteins in organisms living in energy-limiting and extreme environmental conditions. Therefore, our results provide a means for understanding the diversity and significance of proteostasis mechanisms in extreme acidophilic bacteria.
Copyright: © 2023 Izquierdo-Fiallo et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors declare that there are no competing interests.
Figures





Similar articles
-
Adaptive response of the holdase chaperone network of Acidithiobacillus ferrooxidans ATCC 23270 to stresses and energy sources.World J Microbiol Biotechnol. 2025 Apr 1;41(4):121. doi: 10.1007/s11274-025-04325-7. World J Microbiol Biotechnol. 2025. PMID: 40167894
-
Genomic insights into microbial iron oxidation and iron uptake strategies in extremely acidic environments.Environ Microbiol. 2012 Jul;14(7):1597-611. doi: 10.1111/j.1462-2920.2011.02626.x. Epub 2011 Nov 3. Environ Microbiol. 2012. PMID: 22050575 Review.
-
Distribution of Acidophilic Microorganisms in Natural and Man-made Acidic Environments.Curr Issues Mol Biol. 2021;40:25-48. doi: 10.21775/cimb.040.025. Epub 2020 Mar 11. Curr Issues Mol Biol. 2021. PMID: 32159522
-
Carbon, iron and sulfur metabolism in acidophilic micro-organisms.Adv Microb Physiol. 2009;54:201-55. doi: 10.1016/S0065-2911(08)00003-9. Adv Microb Physiol. 2009. PMID: 18929069 Review.
-
Metabolic diversity and adaptive mechanisms of iron- and/or sulfur-oxidizing autotrophic acidophiles in extremely acidic environments.Environ Microbiol Rep. 2016 Oct;8(5):738-751. doi: 10.1111/1758-2229.12435. Epub 2016 Jul 7. Environ Microbiol Rep. 2016. PMID: 27337207
Cited by
-
Osmotic response in Leptospirillum ferriphilum isolated from an industrial copper bioleaching environment to sulfate.Front Microbiol. 2024 May 24;15:1369244. doi: 10.3389/fmicb.2024.1369244. eCollection 2024. Front Microbiol. 2024. PMID: 38855770 Free PMC article.
-
Surfing in the storm: how Paraburkholderia xenovorans thrives under stress during biodegradation of toxic aromatic compounds and other stressors.FEMS Microbiol Rev. 2025 Jan 14;49:fuaf021. doi: 10.1093/femsre/fuaf021. FEMS Microbiol Rev. 2025. PMID: 40388301 Free PMC article. Review.
-
Acidithiobacillia class members originating at sites within the Pacific Ring of Fire and other tectonically active locations and description of the novel genus 'Igneacidithiobacillus'.Front Microbiol. 2024 Apr 3;15:1360268. doi: 10.3389/fmicb.2024.1360268. eCollection 2024. Front Microbiol. 2024. PMID: 38633703 Free PMC article.
-
Adaptive response of the holdase chaperone network of Acidithiobacillus ferrooxidans ATCC 23270 to stresses and energy sources.World J Microbiol Biotechnol. 2025 Apr 1;41(4):121. doi: 10.1007/s11274-025-04325-7. World J Microbiol Biotechnol. 2025. PMID: 40167894
-
Comparative whole genome analysis of face-derived Streptococcus infantis CX-4 unravels the functions related to skin barrier.Genes Genomics. 2024 Apr;46(4):499-510. doi: 10.1007/s13258-024-01495-w. Epub 2024 Mar 7. Genes Genomics. 2024. PMID: 38453815
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
