

Genetic manipulation of Patescibacteria provides mechanistic insights into microbial dark matter and the epibiotic lifestyle
- PMID: 37683634
- PMCID: PMC10633639
- DOI: 10.1016/j.cell.2023.08.017
Genetic manipulation of Patescibacteria provides mechanistic insights into microbial dark matter and the epibiotic lifestyle
Abstract
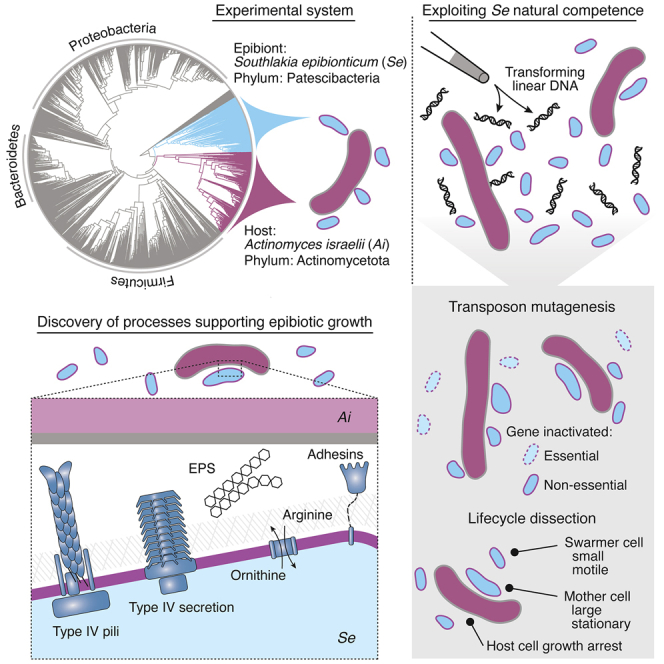
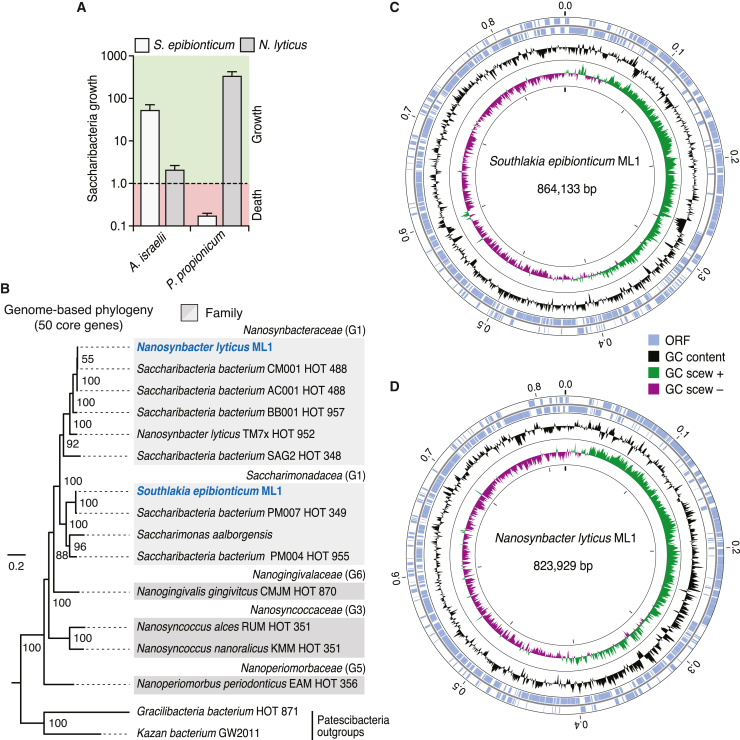
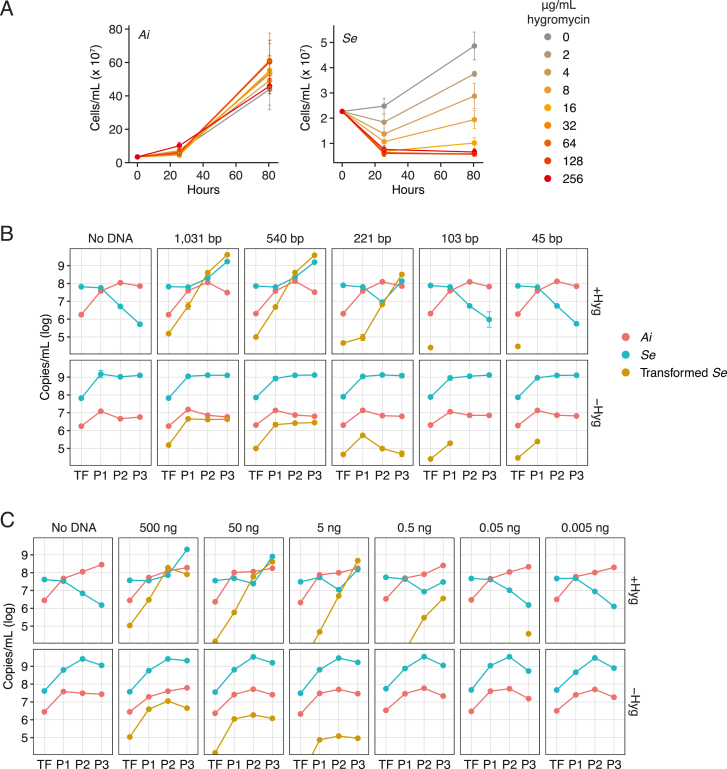
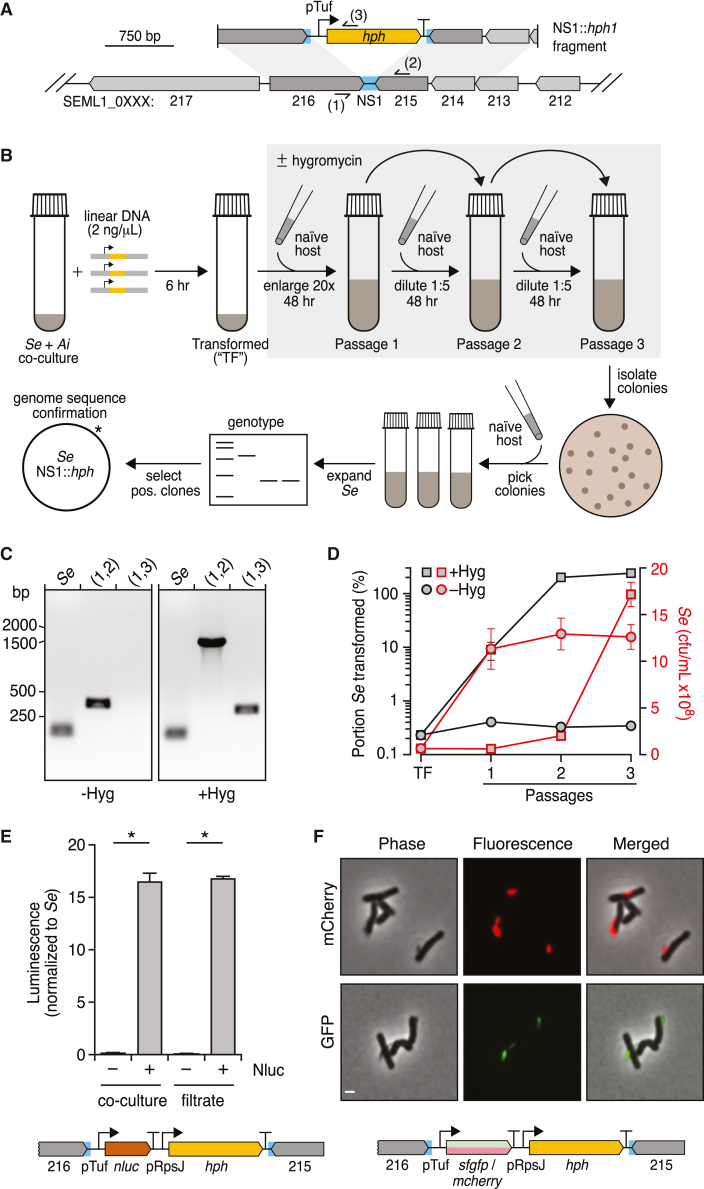
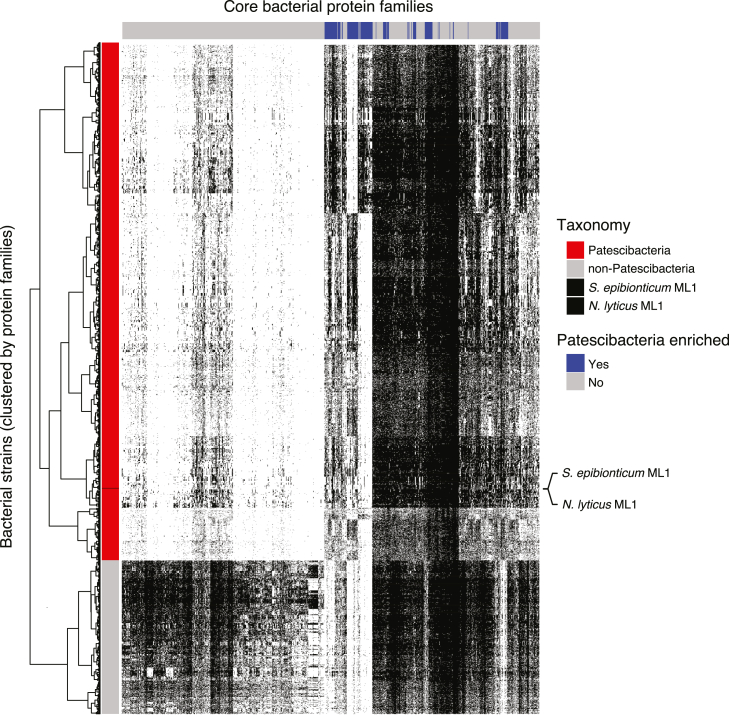
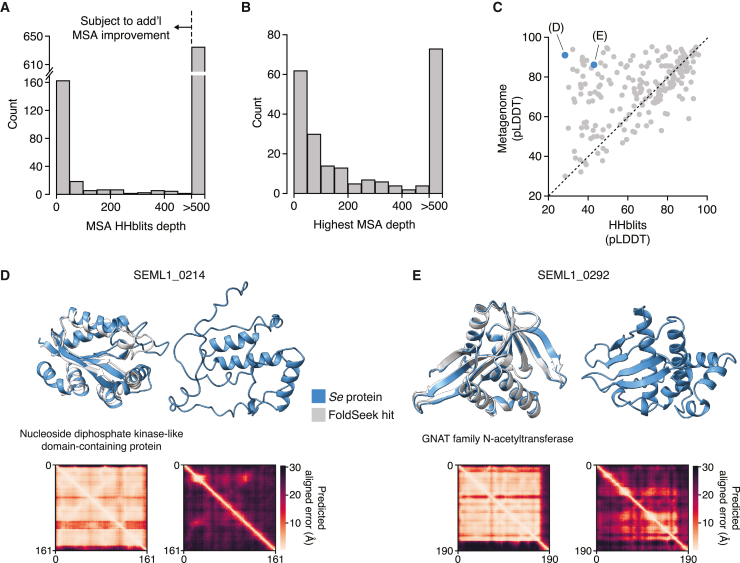
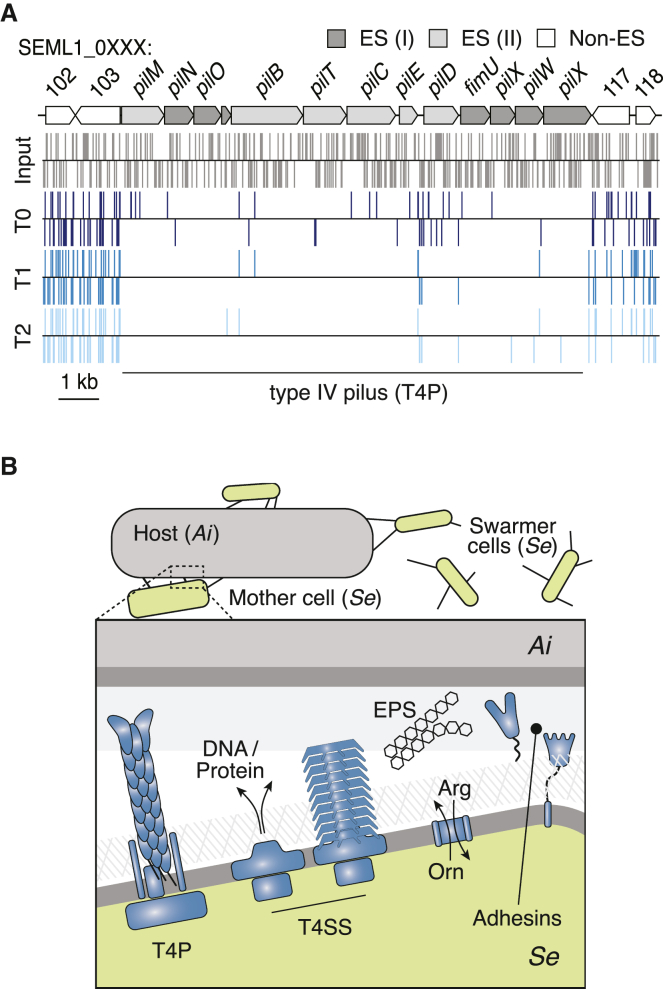
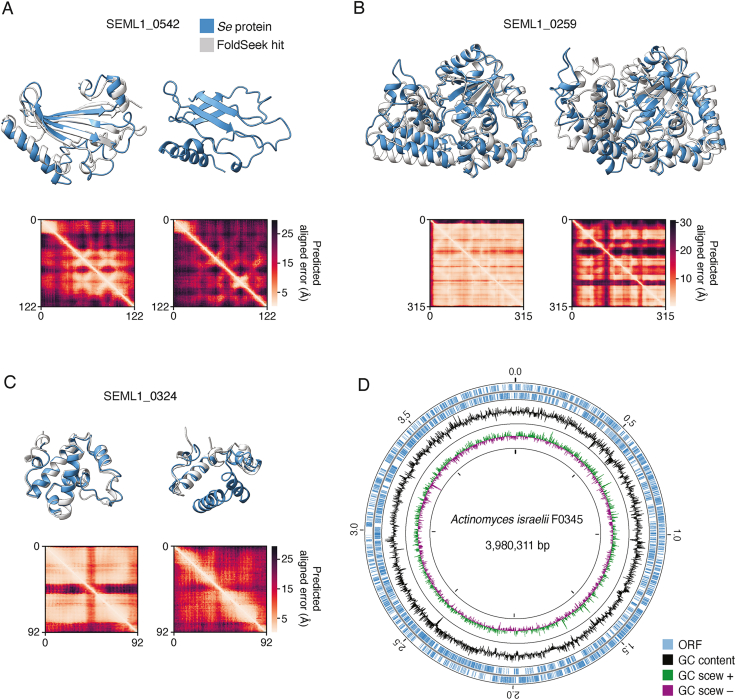
Patescibacteria, also known as the candidate phyla radiation (CPR), are a diverse group of bacteria that constitute a disproportionately large fraction of microbial dark matter. Its few cultivated members, belonging mostly to Saccharibacteria, grow as epibionts on host Actinobacteria. Due to a lack of suitable tools, the genetic basis of this lifestyle and other unique features of Patescibacteira remain unexplored. Here, we show that Saccharibacteria exhibit natural competence, and we exploit this property for their genetic manipulation. Imaging of fluorescent protein-labeled Saccharibacteria provides high spatiotemporal resolution of phenomena accompanying epibiotic growth, and a transposon-insertion sequencing (Tn-seq) genome-wide screen reveals the contribution of enigmatic Saccharibacterial genes to growth on their hosts. Finally, we leverage metagenomic data to provide cutting-edge protein structure-based bioinformatic resources that support the strain Southlakia epibionticum and its corresponding host, Actinomyces israelii, as a model system for unlocking the molecular underpinnings of the epibiotic lifestyle.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
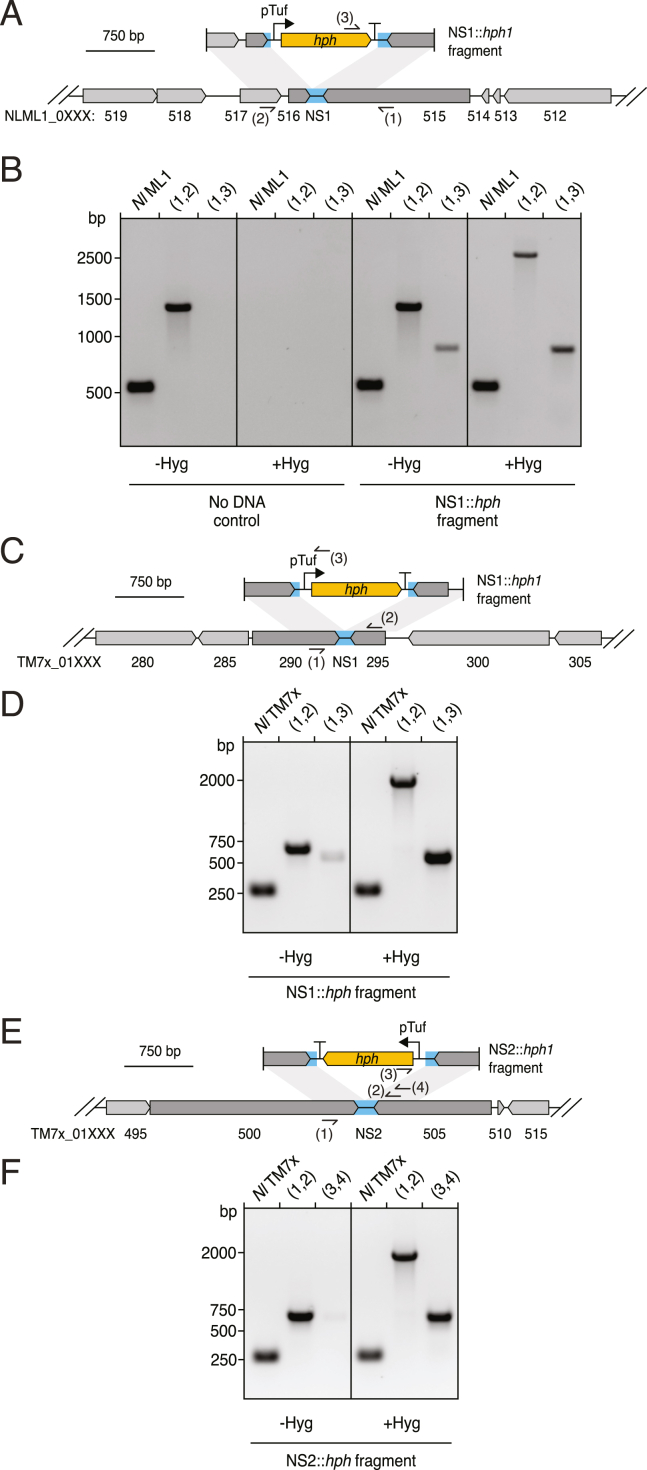
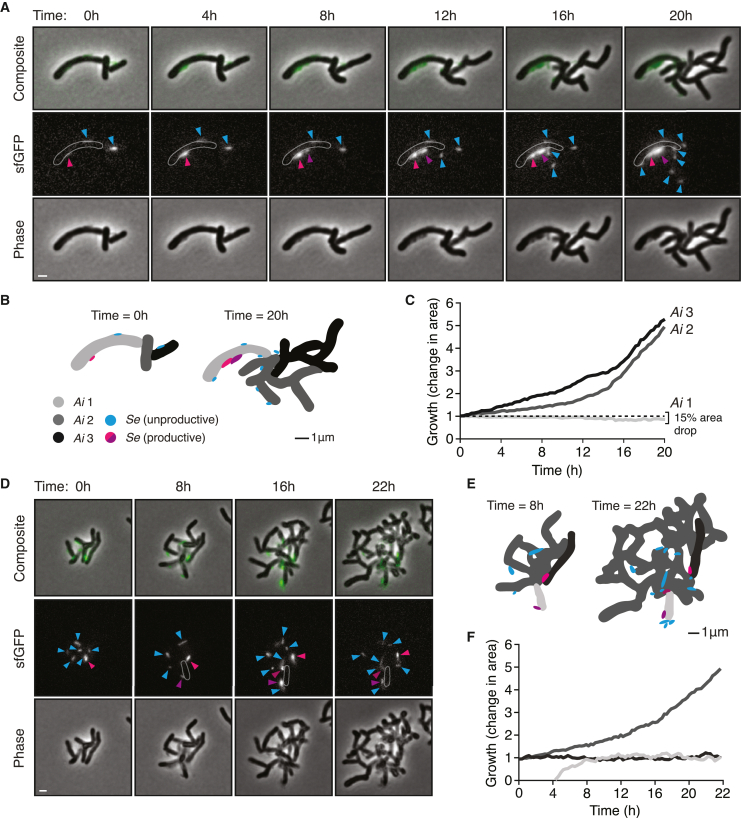
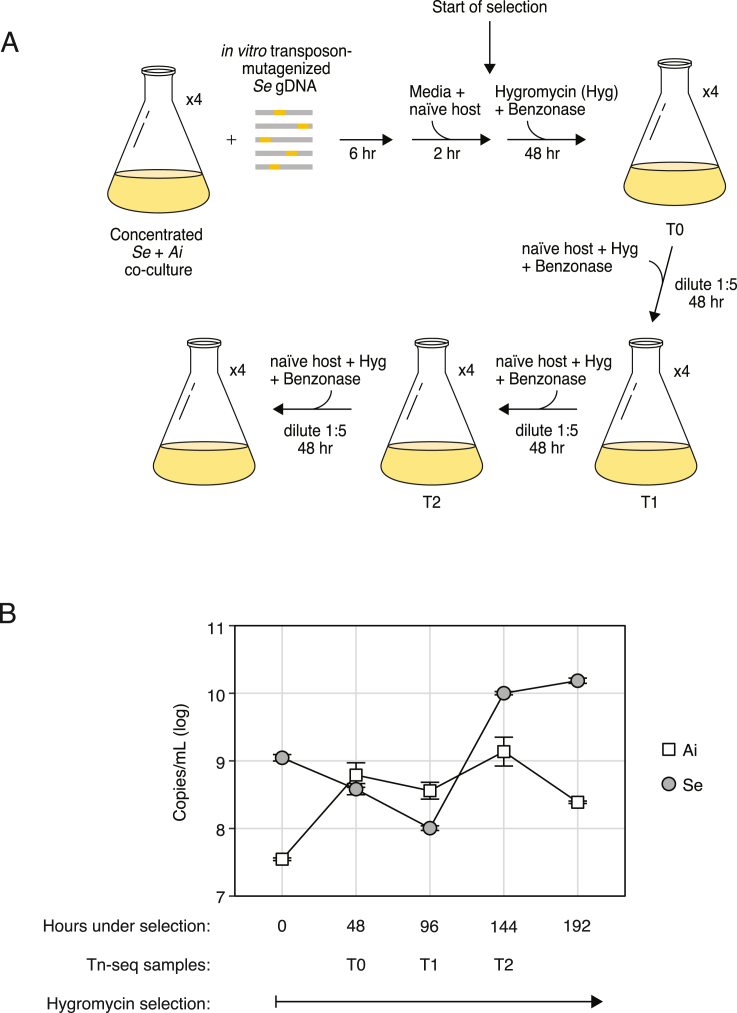
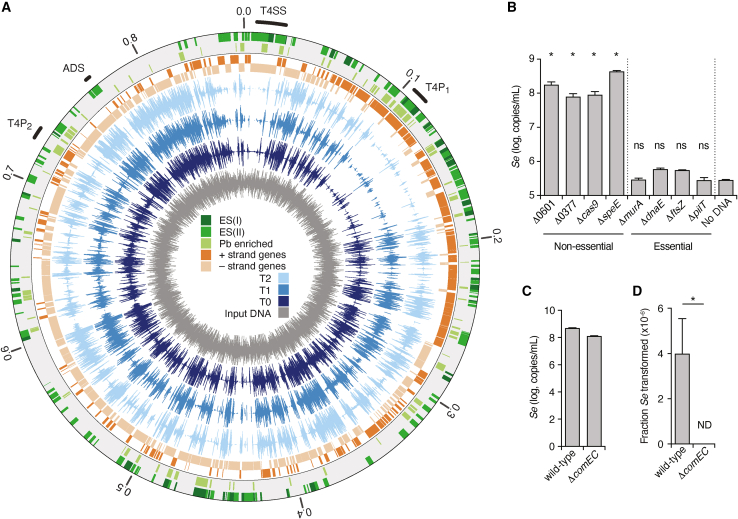
Update of
-
Genetic manipulation of candidate phyla radiation bacteria provides functional insights into microbial dark matter.bioRxiv [Preprint]. 2023 May 11:2023.05.02.539146. doi: 10.1101/2023.05.02.539146. bioRxiv. 2023. Update in: Cell. 2023 Oct 26;186(22):4803-4817.e13. doi: 10.1016/j.cell.2023.08.017. PMID: 37205512 Free PMC article. Updated. Preprint.
References
-
- Marcy Y., Ouverney C., Bik E.M., Lösekann T., Ivanova N., Martin H.G., Szeto E., Platt D., Hugenholtz P., Relman D.A., Quake S.R. Dissecting biological "dark matter" with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc. Natl. Acad. Sci. USA. 2007;104:11889–11894. doi: 10.1073/pnas.0704662104. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
